
Cuproptosis:銅がもたらす諸刃の作用
Jeyashree Alagarsamy著(Cincinnati大学、PhD Candidate)
銅(Cu)は、ミトコンドリアの呼吸系、抗酸化による防御機構、多数の生体化合物の産生等の多岐にわたる生理機能において極めて重要な役割を果たす必須の微量栄養素です。しかし、過剰な銅イオンは有害となり得るため、体内ではその濃度が精緻に調節されています。近年の研究で、Cuproptosis(キュプロトーシス/キュプロプトーシス)と呼ばれる新規の銅依存性細胞死機構が明らかになっています。このCuproptosisは、アポトーシス(Apoptosis)、フェロトーシス(Ferroptosis)、ネクロトーシス(Necroptosis)のような既知の細胞死経路とは著しく異なる細胞死機構です。本稿では、銅イオンの恒常性維持機構や、制御不全が疾患を引き起こす仕組み、Cuproptosisが果たす可能性のある役割について解説します。
|
目次 細胞内の銅イオンの恒常性維持機構 |
細胞内の銅イオンの恒常性維持機構
細胞内の銅イオンの恒常性は、銅結合酵素、銅シャペロン、細胞膜トランスポーターで構成される銅依存性タンパク質の複雑なネットワークによって緊密に制御されています。これらのタンパク質は協調して銅イオンの取込み・排出・細胞内における銅イオンの利用をマネジメントすることで、細胞内の銅イオン濃度を所定の範囲に維持する働きを示し、銅の過剰な蓄積や欠乏による有害な作用を防ぐうえで重要な役割を果たします。細胞内への銅イオンの取込みは、高親和性銅輸送タンパク質CTR1(遺伝子:SLC31A1)がその大部分を担い、1価の銅イオン(Cu+)が細胞内に流入します。CTR1の発現は銅イオンの存在量によって変化し、銅イオンが欠乏している時はアップレギュレーションし、過剰に存在している時はダウンレギュレーションします1,2。CTR1の機能は、銅イオンの腸管吸収において極めて重要な役割を果たし、CTR1の欠乏は末梢臓器の銅イオンの枯渇を引き起こします3。また、ATOX1(Antioxidant-1)とCCS(Copper Chaperone for Superoxide dismutase)は細胞内に銅イオンを輸送するシャペロンタンパク質です。ATOX1は、トランスゴルジネットワーク(Trans-Golgi network)に存在する銅依存性ATPase、ATP7AとATP7Bへ銅イオンを輸送し、銅結合酵素に銅イオンを供給します。一方、CCSはスーパーオキシドジスムターゼ1(SOD1:Superoxide dismutase 1)へ銅イオンを輸送し、SOD1は取り込んだ金属イオンを利用して活性酸素種(ROS:reactive oxygen species)の一種であるスーパーオキシドアニオン(O2-・)を不均化除去します。細胞内の銅イオンの排出はATP7AとATP7Bによって制御され、細胞内の銅イオン濃度が上昇すると、ATP7AとATP7Bはトランスゴルジネットワークから小胞を経由して細胞膜へ移動して過剰な銅を排出します。このようなATPaseが変異するとMenkes病(Menkes disease、ATP7A変異)やWilson病(Wilson's disease、ATP7B変異)のような疾病を発症するという事実は、銅イオンの恒常性の重要性を浮き彫りにしています。
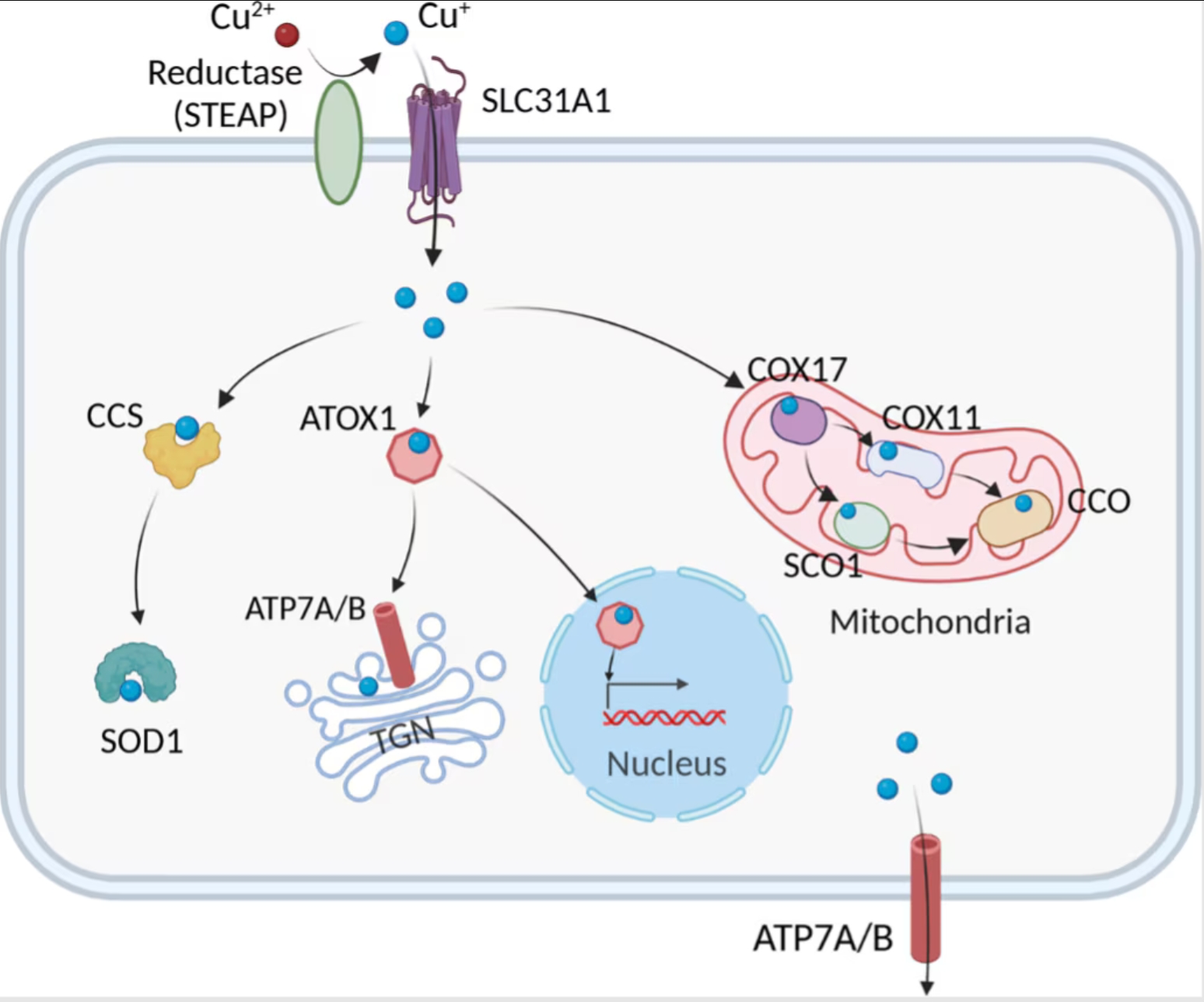
図1. 細胞内の銅イオン代謝経路(Chen, et al. 2022)
|
|
|
|
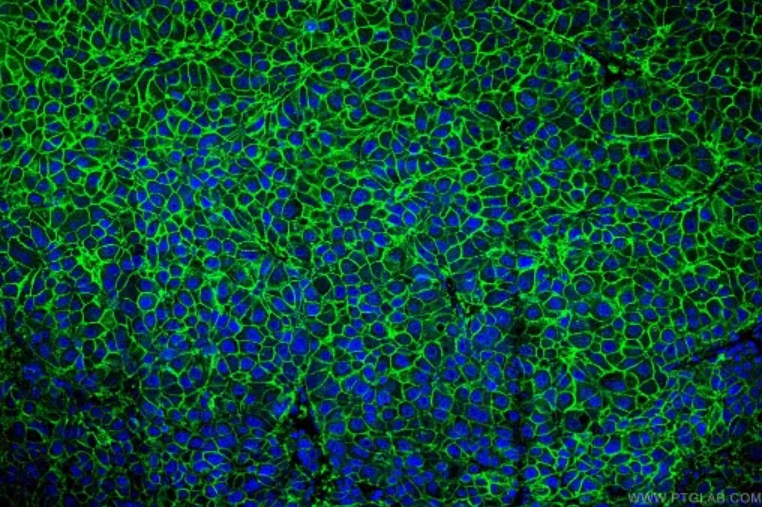
図2. SLC31A1抗体(カタログ番号:67221-1-Ig)、CoraLite488標識AffiniPure Goat Anti-Mouse IgG(H+L)抗体を使用したヒト肝臓がん組織の免疫蛍光染色解析像。 |
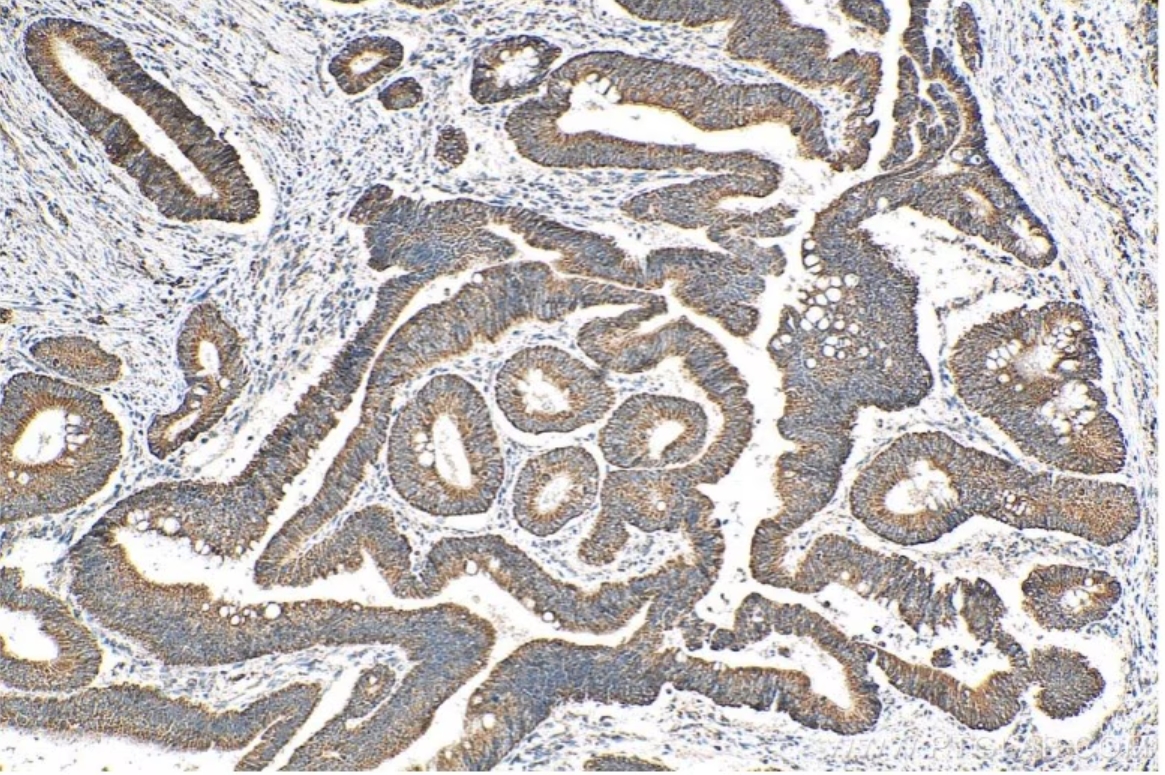
図3. ATP7B-Specific抗体(カタログ番号:19786-1-AP)を使用したパラフィン包埋ヒト結腸がん組織の免疫組織化学染色。Tris-EDTA buffer(pH 9.0)で熱処理し抗原賦活化した試料を使用。 |
Cuproptosisの間、細胞に何が起きているのか?
Cuproptosis発生時の細胞内では、いくつかの顕著な生化学的・形態学的変化が生じます。細胞内に銅イオンが蓄積すると、ROSの産生増加と酸化ストレスが引き起こされ、脂質、タンパク質、DNAのような細胞を構成する物質にダメージを与えます。また、銅イオンがトリカルボン酸回路(TCA回路:Tricarboxylic acid cycle)に関与するタンパク質の異常なリポイル化やタンパク質凝集を促進することで、ミトコンドリア機能が減弱します4,5。さらに、鉄硫黄(Fe-S)クラスタータンパク質の不安定化によって電子伝達機能と酵素活性が阻害され、ミトコンドリア呼吸鎖の反応が滞ります。銅イオンは、ユビキチン‐プロテアソーム系も阻害し、損傷タンパク質の蓄積をまねくとともに細胞内ストレスが増大します。タンパク質凝集やFe-Sクラスタータンパク質の不安定化は、ミトコンドリアの膨張やクリステ構造の消失といった深刻な構造上の損傷を引き起こします。酸化ストレスと脂質過酸化によって透過性が亢進した細胞膜は破裂し、細胞機能の撹乱と損傷した細胞小器官の蓄積が原因となって細胞質に空胞が形成されます6,7。
Cuproptosisの定義と主要な役割を果たす因子
Cuproptosisは、過剰なCu2+イオンが引き金となって生じる制御された細胞死の新規形態であり、アポトーシス、フェロトーシス、ネクロトーシスといったよく知られた細胞死経路とは異なる特徴を示します。Cuproptosisはミトコンドリア酵素の変化と密接な関係があり、ミトコンドリアが銅誘導性細胞死の主要なターゲットとなります8,9。銅イオンへの過剰な暴露はミトコンドリア膜の酸化ダメージの原因となり、TCA回路のタンパク質機能が損なわれます。銅過剰症の患者の場合、TCA回路で重要な役割を果たす酵素アコニターゼの不活性化が、シトクロムcオキシダーゼ(CCO:Cytochrome c oxidase)活性の低下やTCA回路の阻害に関係しています。銅イオノフォア化合物で処理した細胞の代謝産物のプロファイルを調査したところ、TCA回路に関連する代謝産物の調節不全が経時的に生じることが実証されるとともに、電子伝達呼吸鎖複合体Ⅰ・Ⅱを阻害すると銅誘導性細胞死が著しく減弱しました7。DLAT(Dihydrolipoamide S-Acetyltransferase)、FDX1(Ferredoxin 1)、LIAS(Lipoic Acid Synthetase)は、Cuproptosisを制御するうえで鍵を握る重要な遺伝子です。DLATは、ピルビン酸デヒドロゲナーゼ複合体(PDC:Pyruvate dehydrogenase complex)の主要な構成サブユニットで、ピルビン酸由来のアセチル基をCoAに転移してアセチルCoAを生成し、TCA回路に取り込む働きに関与します。一連の働きを示すには、DLATがリポイル化されている必要があります10,11。Cuproptosisが生じる間、銅イオン濃度の上昇によってDLATの異常なリポイル化と凝集が引き起こされ、ミトコンドリアは機能不全に陥って細胞死が生じます。FDX1は、いくつかの代謝経路において電子の伝達に必要なタンパク質であり、Fe-Sクラスターが還元される際に電子を供与します。さらに、FDX1は銅イオンの恒常性と酸化還元プロセスを制御します。この2つの働きは銅誘導性ストレスに対する細胞応答において重要な役割を担います。リポ酸の生合成に必要とされるLIASは、DLATのようなTCA回路の酵素の安定性と活性を維持する働きを示します。LIAS機能の破綻はリポイル化タンパク質の異常凝集の原因となり、このタンパク質凝集がCuproptosisの遠因となります。このような一連の遺伝子は、細胞の生存における銅イオン代謝とミトコンドリア機能の複雑な相互関係を示しています。銅療法はリポイル化が関係する代謝酵素に働きかけ、DLAT、FDX1、LIAS等のTCA回路に関連するタンパク質に影響を及ぼします。これらのタンパク質は、その適切なリポイル化や代謝レベルの維持回復が、銅誘導性細胞死の緩和に極めて重要な役割を果たします12-14。
 |
 |
|
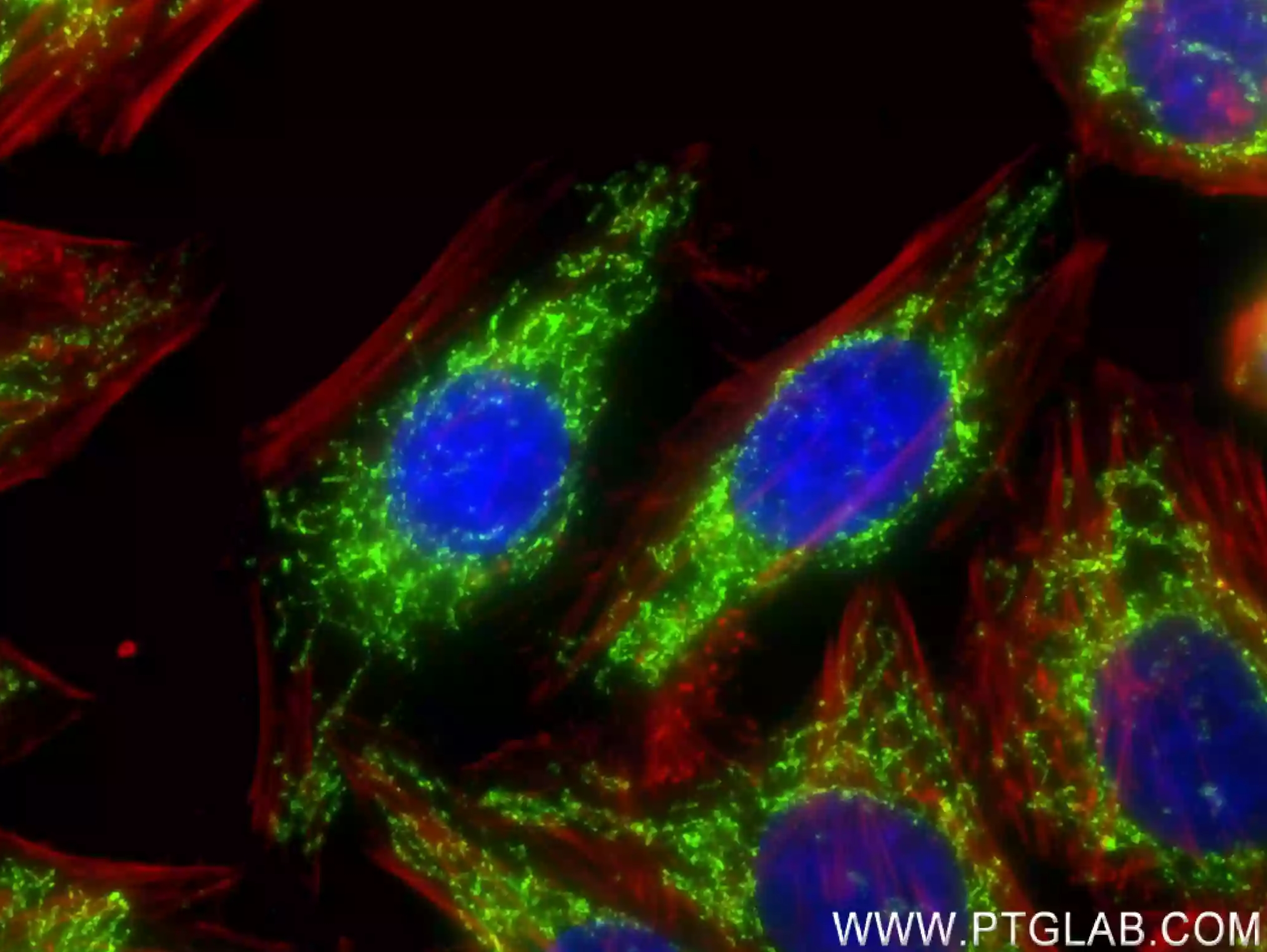
図4. HepG2細胞の免疫蛍光染色解析像。緑:DLAT抗体(カタログ番号:83654-3-RR)/CoraLite®488標識Goat Anti-Rabbit IgG(H+L)抗体(カタログ番号:SA00013-2)。赤:CoraLite®594標識ファロイジン。 |
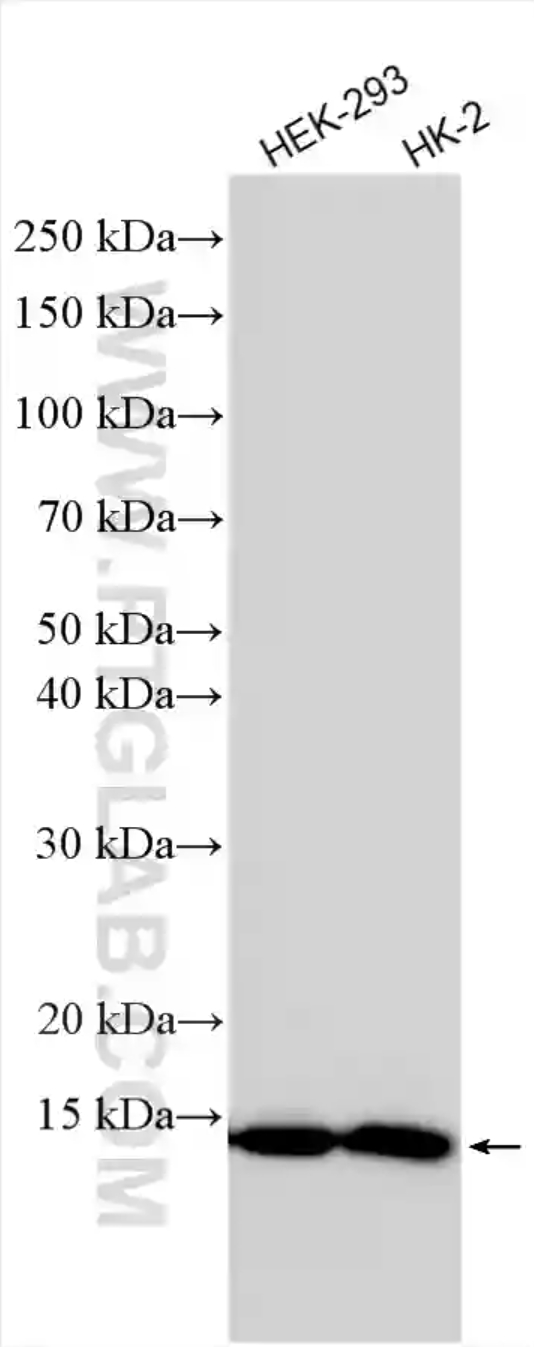
図5. 細胞ライセートのウェスタンブロット。各細胞ライセートをSDS-PAGE後、FDX1抗体(カタログ番号:82957-1-RR)を使用して、室温で1.5時間インキュベーションした。 |
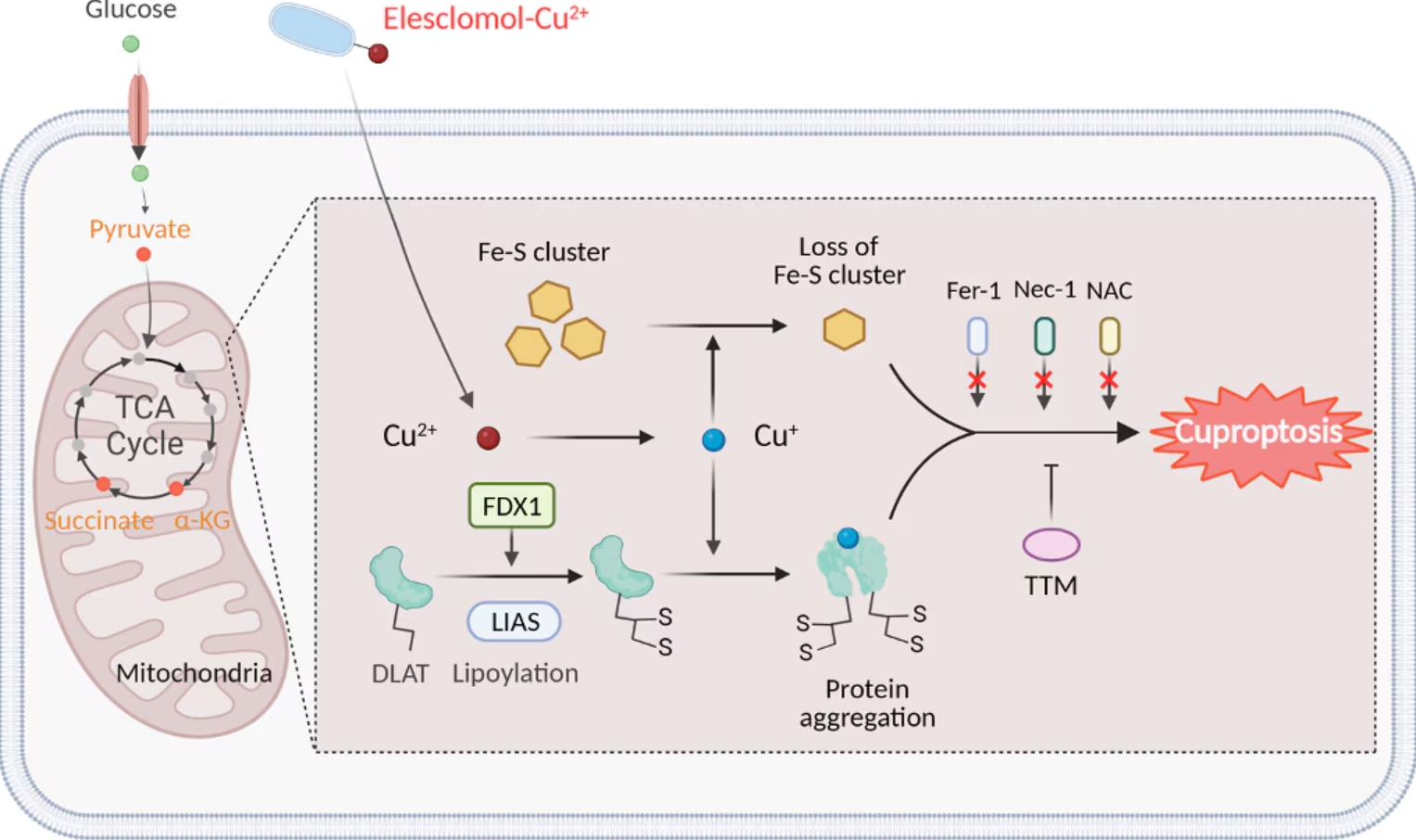
図6. Cuproptosisの模式図(Chen, et al. 2022)
疾患におけるCuproptosisの意義と治療の展望
Wilson病(Wilson's Disease)
Wilson病(WD:Wilson's Disease)はATP7B遺伝子の変異が原因となり発症する疾患で、ATP7Bの機能異常によって脳や肝臓等の組織に銅イオンが蓄積し、深刻な障害を引き起こします。肝臓に関連する症状としては、嘔吐、倦怠感、腹部への体液蓄積(腹水)、下腿腫脹(浮腫)、黄疸(皮膚が黄色くなる)、痒み等が挙げられます。神経症状としては、振戦、筋硬直、発話困難、人格変化、不安障害、幻聴・幻覚が挙げられます15。一般的に、治療は銅吸収を抑制するための経口亜鉛製剤や、ペニシラミン(D-penicillamine)やトリエンチン塩酸塩(Trientine hydrochloride)等のキレート剤を服用することで行います。こうした投薬治療は肝臓症状を緩和しますが、亜鉛製剤やキレート剤の神経症状に対する作用には個人差があります。臨床試験では、テトラチオモリブデン酸(TTM:Tetrathiomolybdate)やビスコリンTTMのような化合物はWilson病患者の神経症状の悪化を緩和できることが示され、2024年時点で試験が継続しています。TTMは銅イオノフォア化合物の有害な作用を阻害することから、Wilson病の進行にはCuproptosisが寄与していると考えられます。α-リポ酸(Cuproptosisに関与する代謝分子)のようなCuproptosisのターゲットとなり得る化合物は、Wilson病の治療薬候補となる可能性が示されています16。Cuproptosisをターゲットとする新薬候補化合物がこの難病の治療に有効性を示すか否かを調査すれば、興味深い結果が得られると考えられます。
神経疾患
Cuproptosisは、アルツハイマー病(AD:Alzheimer's disease)、ハンチントン病(HD:Huntington's disease)、筋萎縮性側索硬化症(ALS:Amyotrophic lateral sclerosis)等のいくつかの神経変性疾患と関連しています17。過剰な銅の存在は疾患の進行に関連し、銅の蓄積がアルツハイマー病やハンチントン病の有害なタンパク質凝集体の形成と関連することが研究で示されています。さらに、銅結合タンパク質であるSOD1の遺伝子変異は、ALSと関連があるとされています。ミトコンドリア機能不全は、銅イオンによる神経毒性における重要な機構であると考えられ、Cuproptosisが一連の神経変性疾患の治療ターゲットとなる可能性があることを示唆しています8。神経変性疾患におけるCuproptosisの役割や、治療薬を開発するうえでCuproptosisが有効な治療ターゲットとなり得るか否かを解明するには、さらに研究を進める必要があります。
がん
Cuproptosisは、がんの治療ターゲット候補として有望視されています。銅イオンはアポトーシスやROSの蓄積を介してがん細胞の細胞死を引き起こす引き金となることが、研究で示されています18。新規のがん治療の手段としては、非多孔性銅配位ポリマーGOx@[Cu(tz)]のようなナノ粒子を採用する手法が挙げられます19。この手法は、動物モデルで腫瘍の増殖を抑制すると同時に毒性を最小限に抑えることが実証されています。エレスクロモール(Elesclomol)のような銅イオノフォア化合物も、ROS産生の増加を介した抗がん作用を示します。エレスクロモールと化学療法を併用する臨床試験では、ミトコンドリアの活性状態が高いことを表す、乳酸脱水素酵素(LDH)の値が低い患者において、特に高い効果を示すという結果が得られています20(※なお、この臨床試験はその後の第Ⅲ相で中止となっています)。Cuproptosisを対象としたがんの治療薬の有効性を確認するには、さらなる研究や臨床試験の実施が必要とされます。
最後に
Cuproptosisの発見は銅イオンの制御と疾患の病態形成との間の複雑な関係性を明らかにしました。Wilson病や神経変性疾患、がんにいたるまで、過剰な銅の存在は細胞にとって有害な機構を引き起こす可能性があり、Cuproptosisが治療のターゲットとなる可能性を示唆しています。Cuproptosisは疾病の治療に有望な機構ではあるものの、様々な疾患におけるCuproptosisを対象とした治療効果を十分に探究するには、より詳細な研究や臨床試験の実施が求められます。
参考文献
- J McDonough, S Sternbach. Effects of cuprizone on mitochondria. Mitochondrial Intoxication. 2023:439-450.
- ZD Liang, et al. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol. 2012 Mar;81(3):455-64.
- Y Nose, et al. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006 Sep;4(3):235-44.
- M Wang, et al. Cuproptosis: emerging biomarkers and potential therapeutics in cancers. Front Oncol. 2023 Nov 7:13:1288504.
- MB Dreishpoon, NR Bick, et al. FDX1 regulates cellular protein lipoylation through direct binding to LIAS. J Biol Chem. 2023 Sep;299(9):105046.
- D Tang, G Kroemer, R Kang. Targeting cuproplasia and cuproptosis in cancer. Nat Rev Clin Oncol. 2024 May;21(5):370-388.
- P Tsvetkov, TR Golub, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022 Mar 18;375(6586):1254-1261.
- CT Sheline, DW Choi. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004 May;55(5):645-53.
- M Arciello, et al. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem Biophys Res Commun. 2005 Feb 11;327(2):454-9.
- S-R Li, et al. Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduct Target Ther. 2022 May 13;7(1):158.
- M Ni, RJ DeBerardinis, et al. Functional Assessment of Lipoyltransferase-1 Deficiency in Cells, Mice, and Humans. Cell Rep. 2019 Apr 30;27(5):1376-1386.e6.
- EA Rowland, CK Snowden, IM Cristea. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. 2018 Feb:42:76-85.
- D Brancaccio, et al. [4Fe-4S] Cluster Assembly in Mitochondria and Its Impairment by Copper. J Am Chem Soc. 2017 Jan 18;139(2):719-730.
- S G-Santamarina, MA Uzarska, et al. Cryptococcus neoformans Iron-Sulfur Protein Biogenesis Machinery Is a Novel Layer of Protection against Cu Stress. mBio. 2017 Oct 31;8(5):e01742-17.
- A Ala, et al. Wilson's disease. Lancet. 2007 Feb 3;369(9559):397-408.
- J Smirnova, P Palumaa, et al. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. α-Lipoic acid as a potential anti-copper agent. Sci Rep. 2018 Jan 23;8(1):1463.
- G Gromadzka, et al. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci. 2020 Dec 4;21(23):9259.
- A Gupte, RJ Mumper. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009 Feb;35(1):32-46.
- Y Xu, L Zeng, Y He, Z Dai, Y Pan, et al. An Enzyme-Engineered Nonporous Copper(I) Coordination Polymer Nanoplatform for Cuproptosis-Based Synergistic Cancer Therapy. Adv Mater. 2022 Oct;34(43):e2204733.
- JR Kirshner, et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008 Aug;7(8):2319-27.

免疫沈降用アフィニティビース
<Nano-Trap 無料サンプル配布中!>