
フローサイトメトリーパネルの構築ガイド
Frédéric Duval博士、Sari Gezzar-Dandashi著(Maisonneuve-Rosemont病院研究センター)
フローサイトメトリーは1秒間に数千個の細胞を解析し、細胞集団を詳細に精査できる手法で、今や様々な研究分野で欠かせない手法となっています。測定精度を左右するのがフローサイトメトリーパネルで、サンプルとなる細胞のマーカー発現レベルを正確に測定するうえで大きな役割を果たします。課題となるのは、精度が高く実用的なフローサイトメトリーパネルの構築です。本稿では、フローサイトメトリーの初心者の方にもベテラン研究者の方にも役立つ情報を交えつつ、パネル構築のプロセスを解説します。
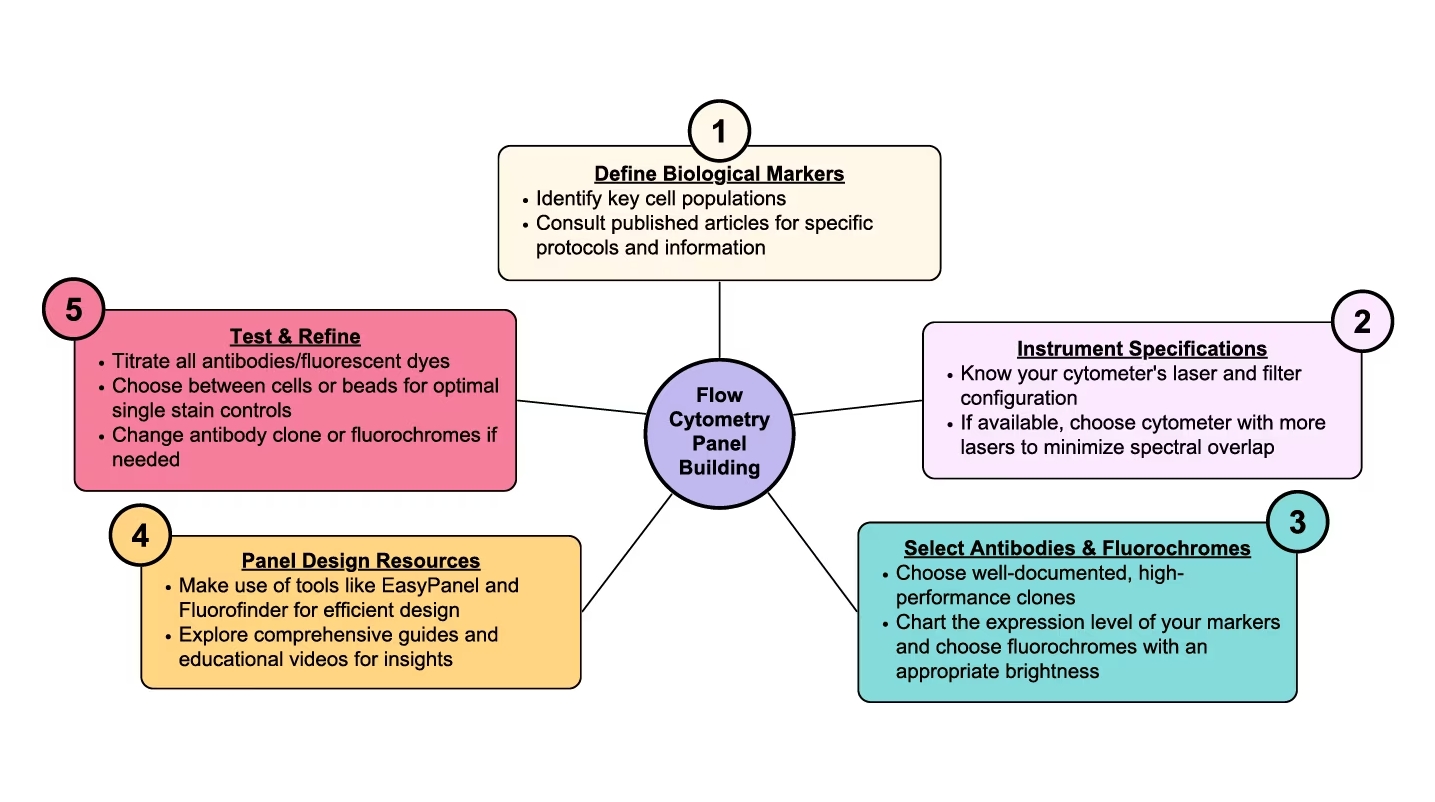
<フローサイトメトリーのパネル構築>
1. 研究課題の解明に必要なマーカーを具体的に決める
2. 装置について理解する
3. 使用する抗体と蛍光色素を選択する
4. パネルデザインのサポートツール
5. パネルの評価・検証・改善
1. 研究課題の解明に必要なマーカーを具体的に決める
系統マーカー(Lineage Marker):系統マーカーは、研究対象の細胞集団を識別するうえで極めて重要になります。例えば、T細胞集団の同定にはCD3マーカーを使用しますが、T細胞集団をCD4+細胞集団(CD3+、CD4+、CD8-)とCD8+細胞集団(CD3+、CD8+、CD4-)に分類するには、CD4マーカーとCD8マーカーを使用します。
除外マーカー(Exclusion Marker):除外マーカーは、解析対象ではない細胞を除外してシグナル分解能を改善するために用います。例えば、末梢血単核細胞(PBMC:Peripheral Blood Mononuclear Cells)中のNK細胞を解析する場合は、T細胞とB細胞を解析から除外するためのT細胞マーカーとB細胞マーカーを追加することがあります(図1)。
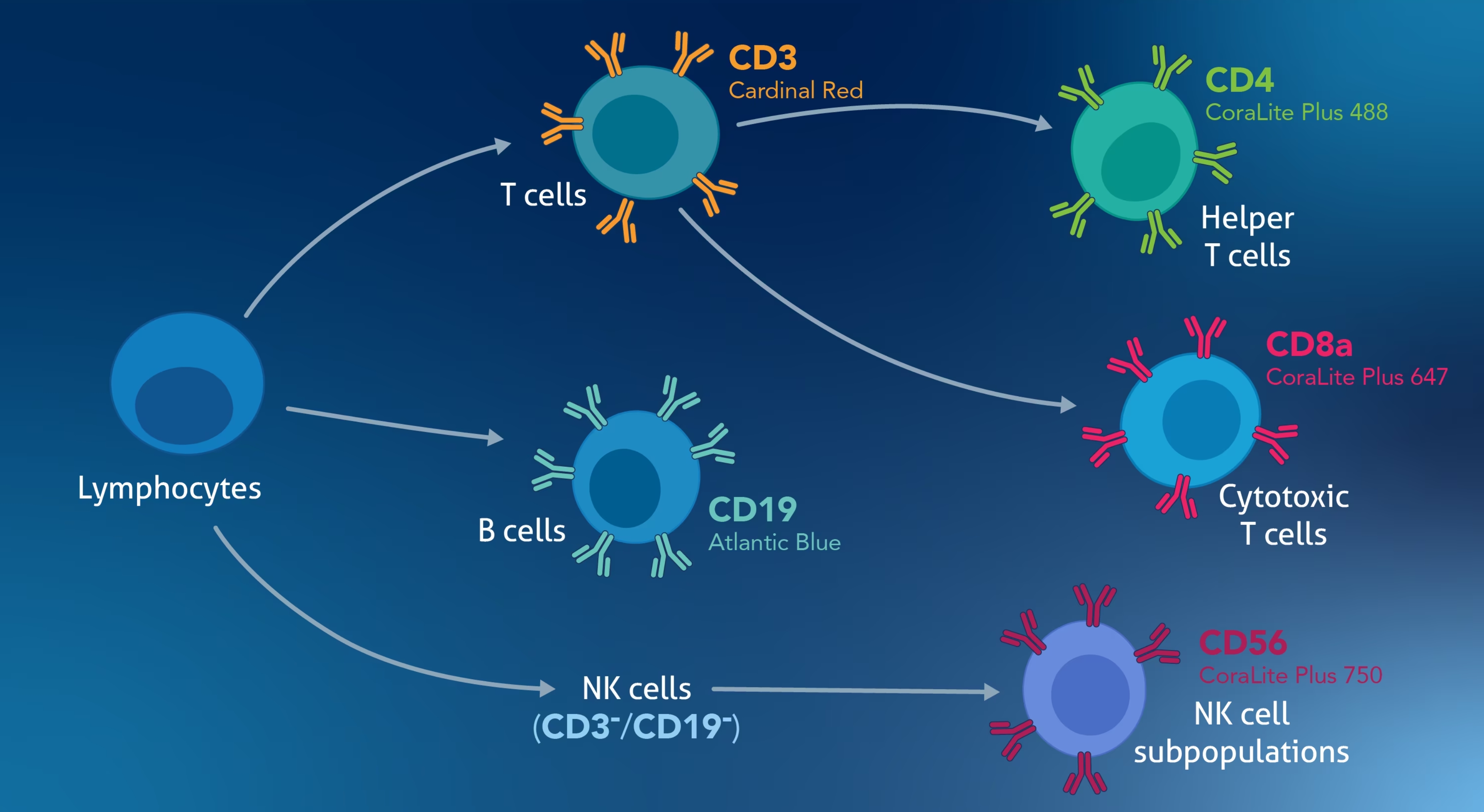
図1. 解析対象の細胞集団がNK細胞の場合、Human TBNK Basics Panel(カタログ番号:PK30050)のようにCD3とCD19を除外マーカーとして使用すると、T細胞とB細胞を解析から除外することができます。
解析対象マーカー :研究課題に直結するマーカーで、活性化マーカー・分化マーカー・DNA結合色素等が解析対象マーカーとして挙げられます。
|
ポイント1:できる限り既存のパネルや知見を利用する Optimized Multicolor Immunophenotyping Panel(OMIP)の論文のような様々なリソースが公開されており、研究対象の細胞集団を検出するうえで参考になるプロトコールを確認したり、他の研究者が利用している様々な抗体クローンを調べることができます。こうした情報は、パネル構築・試薬に関する情報・ゲーティング方法等の情報収集にも役立ちます。 |
2. 装置について理解する
-
フローサイトメーターの検出器の数、光学フィルターの設定、搭載するレーザー数、照射可能な励起波長等を把握しておくことが極めて重要です。装置のレーザー波長の設定で使用する蛍光色素を励起できなければなりません。さらに、蛍光シグナルを精密に捕捉し、結果を正しく解釈するには、蛍光色素の発光スペクトルのピークとフローサイトメーターの光学フィルターが適合している必要があります。機器の設定は機器ごとに異なるため、施設の機器管理者に尋ねてみるかマニュアルを参照しましょう。最終手段として、シリアル番号を確認して製造会社に問い合わせ、使用機器に関する情報を入手することも可能です。
-
スペクトルの重複を最小限に抑えるために、より多くのレーザーを搭載した機器を使用しましょう。搭載レーザー数は、大規模なパネルを使用する場合に、蛍光の漏れ込みを低減し、分解能を向上させるうえで極めて重要となります。
プロテインテックの関連ブログ:フローサイトメトリーのスペクトル補正について -
良好な結果を得られるように、装置の状態を最適に保ち、適切なキャリブレーションを必ず実施してください。
3. 使用する抗体と蛍光色素を選択する
-
論文等への使用実績があり、優れたパフォーマンスを示した抗体クローンを選択しましょう。既知の情報がない場合は、複数の抗体クローンを使用して検証を実施し、陰性の細胞集団と陽性の細胞集団を最も良好に識別できる抗体クローンを選択します(例:染色指数(Staining index)の比較)。さらに、リファレンスとなる細胞集団、既存の文献で検証済みの細胞株、アイソタイプコントロール抗体、またはこうした細胞サンプルとアイソタイプコントロール抗体を併用して、研究対象のサンプルに対するクローン抗体の特異性評価を行います。例えば、ターゲットとなるマーカーをノックアウトした細胞株は陰性コントロールサンプルとして使用できます。
プロテインテックの関連製品:アイソタイプコントロール抗体 -
使用するパネルのマーカーを発現レベルに基づいて分類し、発現量の少ないマーカーや発現量が未知のマーカーにPE等のより明るい蛍光色素を割り当てます。反対に、発現量の多いマーカーには明るくない蛍光色素を割り当てます。蛍光色素とマーカーの組み合わせに配慮することで、サンプル中の様々な細胞集団を識別できると同時に、最良の分解能で対象のマーカーを検出できます。
-
特に共発現マーカーを測定する場合はできる限り、それぞれを異なるレーザー波長で励起可能で、蛍光のスピルオーバー(蛍光の漏れ込み)を最小限に抑えられる蛍光色素の組み合わせを選択します。このアプローチにより、分解能の低下に大きく寄与する、スピルオーバーに起因する拡がりの誤差(Spillover spreading error:Trumpet Effect、ポイント2参照)を低減することができます(図2参照)。Trumpet Effectが発生してしまう蛍光色素の組み合わせは、Spillover Spreading Matrix(SSM)に関する情報を参照すればある程度予測できます。機器担当者に装置特有のSSMの既知情報があるか確認し、Trumpet Effectを回避しましょう。
-
特に、タンデム色素が結合している抗体は、CD8・CD3・CD4のように階層上位の系統マーカーや、高発現する主要なマーカーを検出する際に組み入れないようにします。パネルの別の抗体が、そのタンデム色素のドナーとなる蛍光色素と結合していないか注意しましょう。例えばAPC結合抗体を使用する場合は、階層構造的で多くの細胞に発現するマーカーを検出する目的でAPC-Cy7が結合しているタンデム色素結合抗体を組み入れない方が良いでしょう。その性質上、タンデム色素のドナー色素(APC)からアクセプター色素(Cy7)へのエネルギー移動は不完全であることに加え、色素の劣化・分解等によりエネルギーの一部はアクセプター色素へ移行しません。その結果、ドナー色素のAPCから蛍光が生じ、データ分布の拡がりや擬陽性シグナルが発生する可能性があります5。
|
ポイント2:Spillover spreading error(Trumpet Effect)を最小限に抑える パネルデザインやトラブルシューティングの際に、細胞集団を適切に分離し、影響を最小限に抑えるために、押さえておきたいポイントは3つあります。
蛍光色素は幅広い波長にまたがる蛍光を発するため、複数の蛍光色素がパネルに存在すると、多くの場合、各蛍光色素のスペクトルは重複します1。蛍光スペクトルが重複すると、ある蛍光色素の蛍光が別の蛍光を測定するための検出器に漏れ込みます。目的の波長の蛍光はプライマリー検出器(主検出器)によって捕捉されます。一方で、プライマリー検出器以外の検出器(セカンダリー検出器)が漏れ込んだ蛍光を捕捉します。検出器に漏れ込んだシグナル量を計測して検出した光子の合計から差し引き、蛍光補正(コンペンセーション)と呼ばれる数学的な補正計算を実施して検出したい蛍光色素から生じたと考えられる蛍光を計算します2-5。 しかし、別の蛍光色素由来の蛍光がセカンダリー検出器へ漏れ込むと、感度低下の原因になります。漏れ込んだシグナルは、セカンダリー検出器による光子の計測を不正確なものにします6。Spillover spreading errorは蛍光強度が強いほど大きくなりますが、検出器側の感度・検出効率が低い場合も、相対的にデータの拡がりが増大しやすくなる傾向があります。この傾向は、光電子倍増管(PMT:Photo multiplier tube)で近赤外線側の波長を検出するケースのように、蛍光色素が発する光子の波長範囲が検出器に適していない範囲である場合により強く現れます7。 したがって、コンペンセーション値を根拠にSpillover spreading errorを予測してもその信頼性は限定的です。分解能の低下は陽性シグナルと陰性シグナルを区別する大きな妨げとなります。特に、発現レベルが低く分離が不明瞭なマーカーにおいてその影響は顕著になり8-11、研究対象となるマーカーを検出する際に問題となる場合があります。 |
-
可能な限り、使用する蛍光色素の選定段階で、実際に使用する装置で未染色サンプルを測定してサンプルの自家蛍光現象のパターンを確認します。自家蛍光のシグナルが極めて強い領域と被るチャネルは使用を避けましょう。自家蛍光シグナルが強いと分解能が低下すると予測されるため、もし自家蛍光シグナルの強いチャネルを使用する場合はそのチャネルに明るい蛍光色素を割り当てます。
-
除外マーカーの検出には「ダンプチャネル(Dump channel)」を使用します。この方法では、除外したいマーカーすべてに1種類のダンプチャネル用の蛍光色素を適用します。そうすることで、除外マーカーを発現する解析に不必要な細胞すべてを一括同定して解析から除外できます。
-
データ品質の向上には、死細胞/生細胞識別用の色素を併用します。細胞膜の選択的透過性が失われた死細胞や瀕死の細胞を識別色素で染色し、当該色素によるシグナルを生成する細胞を除外します。
プロテインテックの関連製品:死細胞/生細胞識別色素(Viability Dye)
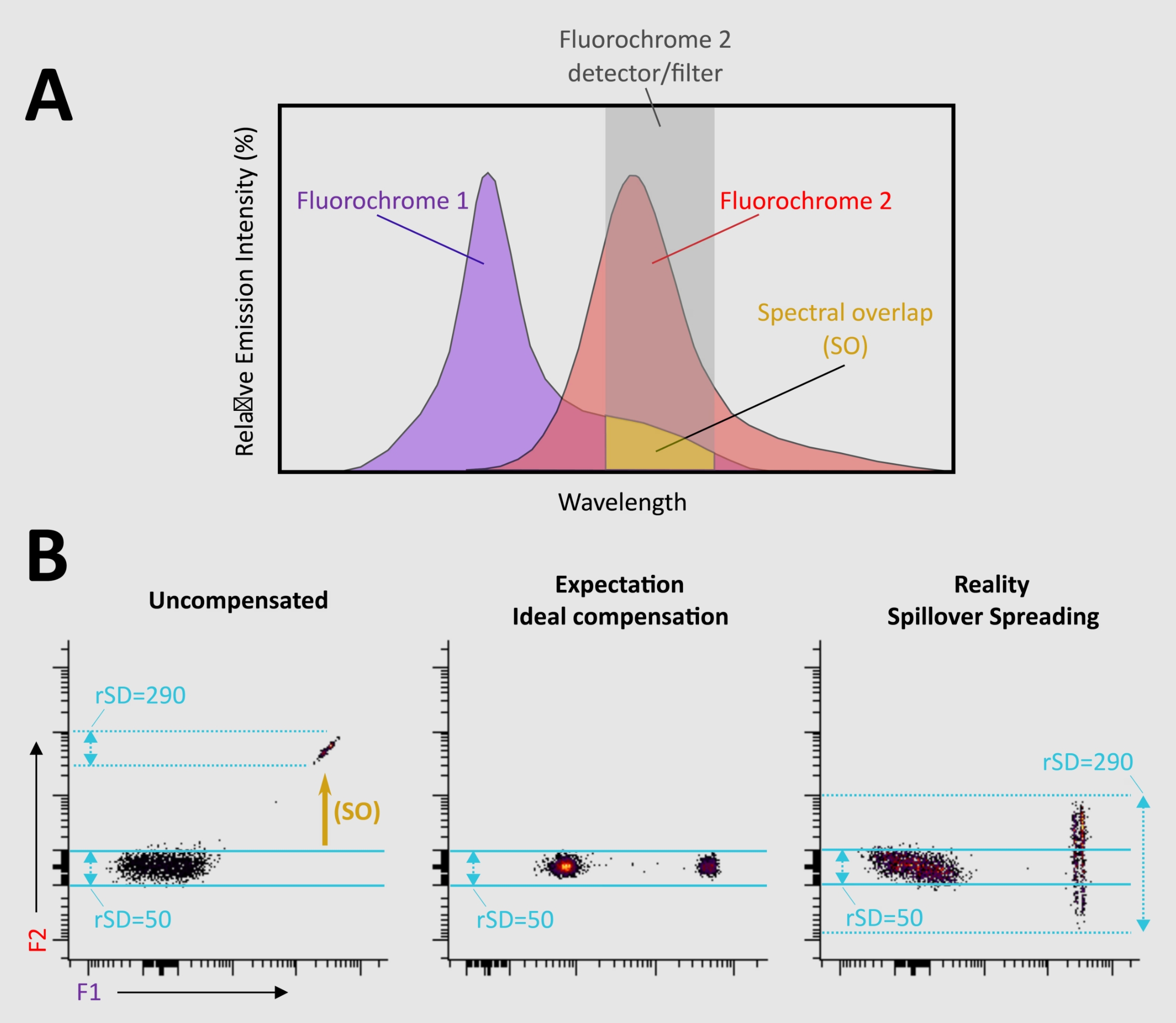
図2. 適切な補正を実施してもSpillover spreading error(Trumpet Effect)は発生する場合があります。A)蛍光色素1(F1)および蛍光色素2(F2)の蛍光スペクトルに、F2検出器に設置した光学フィルターを重ねました。スペクトルの重複(SO)が存在するため、F2検出器はF1由来のシグナルも捕捉します。B)左パネル:(補正前)F1標識抗体だけをインキュベーションしたビーズをフローサイトメーターで測定すると、F2検出器にF1由来の漏れ込みシグナルが観察されます。データ分布のばらつきを示すロバスト標準偏差(rSD:Robust standard deviation)=50の領域にF1陰性集団のプロットが認められ、rSD=290の領域にもF1陽性集団のプロットが生じます。中央パネル:(補正後:Spillover spreading errorのない理想例)F2検出器におけるF1色素の影響は適切に補正されています。サンプル集団にSpillover spreading errorが生じない場合、補正後のF1陽性集団のrSDはF1陰性集団のrSDと同じ値をとり、このパネルではrSD=50となります。右パネル:(補正後:Spillover spreading errorが認められる場合)Spillover spreading error(Trumpet Effect)が発生すると、適切な補正処理を実施した後であっても、F1陽性シグナルがパネル中の青の実線を越え、F2チャンネルの分布が「拡がり」ます(rSD=290)。 その結果、F2で染色したマーカーが「拡がり」の領域と重複する連続的な分布パターンを示す場合や、マーカー自体の発現量が少ない場合は検出が困難になる場合があります。このような事例では、パネルから1つの蛍光標識抗体だけを除いて処理したFMO(Fluorescence minus one)サンプルを測定すると、陰性集団と陽性集団の区別に有用な情報が得られます(図2の場合はF2標識抗体を除外して測定します)。細胞集団の陰性/陽性を正確に区別できない場合は、代わりにSpillover spreading errorが発生しにくい別の蛍光色素の適用を推奨します。
4. パネルデザインのサポートツール
ソフトウェアツール
-
EasyPanel(flow-cytometry.net)やFluorofinder(fluorofinder.com)のようなパネルデザインツールを使用すると、様々な蛍光色素の励起スペクトルや蛍光スペクトルをプロットし、発生する可能性のあるスペクトルの重複を可視化することができます。このような支援ツールは、蛍光色素の明るさに関する情報も提供します。現在では、蛍光色素の組み合わせを提案する人工知能も搭載し、市販されている抗体や、使用する装置固有の設定、解析対象の推定抗原密度等に基づき、蛍光色素の組みあわせを提案してくれます。
オンライン情報
-
多くの企業が、パネル構築に関する詳細な手順を掲載した包括的な手引きを提供しています。AJA Rieger Flow Cytometry、Expert Cytometry、OpenFlow Cytometry、UChicago FlowといったYouTubeチャンネルでは、フローサイトメーターの仕組みや使用法に関する基礎知識を紹介しています。このようなコンテンツはテキストによる手引よりも手軽なビデオ形式で学習効率も高く、初心者にとって最適なリソースとなります。University of Chicago(uchicago.edu)のウェブサイトでは、実験計画、サンプル調製、データ解析に関する情報からブログ、ChUG Podcast、機器のデモにいたるまで幅広い情報を提供しています。
-
マーカー発現やマーカーの共発現レベルに関する情報は、Human Protein Atlas(proteinatlas.org)、Antibodypedia(antibodypedia.com)、抗体販売会社のウェブサイト、公開論文等に掲載されています。
5. パネルの評価・検証・改善
-
抗体の使用量を抑えつつ、バックグラウンドノイズ(非特異的結合)が大きくなりすぎず染色指数が最大となる抗体希釈倍率を決定するために、すべての抗体/蛍光色素の使用量を検討します。
-
試験サンプルに最も適した単一染色コントロールを決定します。コントロールサンプルは、細胞またはビーズのいずれかを選択します。適切に補正できるように、サンプルと同程度の単一染色シグナル、またはサンプルよりも明るい単一染色シグナルで補正を実施します。
プロテインテックの関連ブログ:フローサイトメトリーのスペクトル補正について -
効率化を図るには、パネルを細分化して検討するのではなく、最初から十分に調査して完全なパネルをデザインすることをおすすめします。あるパネルに抗体/蛍光色素を追加すると、望ましくない結果が生じる場合があります。したがって、最初からパネル全体を最適化することで、無駄な時間の消費と労力を抑えられます。
-
適切なコントロールを使用してパネルの検証を実施します。目的の細胞集団を正確に識別できているか確認し、その際の補正コントロールが適切に蛍光の漏れ込みを修正し、Spillover spreading errorがデータ品質に影響を及ぼしていないことを確認します。こうした問題を克服するには、抗体/蛍光色素を変更する必要がある場合もあります。
効果的なフローサイトメトリーパネルを構築するには、装置を十分に理解し、使用するマーカーと蛍光色素をよく吟味して選択する必要があります。本稿では、リソースの活用と戦略的計画の重要性を中心にパネルデザインのプロセスの要点を解説しました。スペクトルの重複や蛍光の漏れ込み等はフローサイトメトリーを実施するうえで避けられない課題ですが、パネルデザインの慎重な構築や継続的な試行錯誤により軽減することができます。オンラインの豊富なツールやリソースを活用すれば、初心者でも必要な知識を補いながら無理なく実践できるでしょう。

参考文献
- SF Sahir, KS Siveen, et al. Development of a 43 color panel for the characterization of conventional and unconventional T-cell subsets, B cells, NK cells, monocytes, dendritic cells, and innate lymphoid cells using spectral flow cytometry. Cytometry A. 2020 Dec 18.
- M Roederer. Compensation in flow cytometry. Curr Protoc Cytom. 2002 Dec:Chapter 1:Unit 1.14.
- M Roederer. Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry. 2001 Nov 1;45(3):194-205.
- Z Maciorowski, et al. Basic Multicolor Flow Cytometry. Curr Protoc Immunol. 2017 Apr 3:117:5.4.1-5.4.38.
- HT Maecker, et al. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry A. 2004 Dec;62(2):169-73.
- R Nguyen, et al. Quantifying spillover spreading for comparing instrument performance and aiding in multicolor panel design. Cytometry A. 2013 Mar;83(3):306-15.
- D Bhowmick, ML Ratliff, et al. A gain and dynamic range independent index to quantify spillover spread to aid panel design in flow cytometry. Sci Rep. 2021 Oct 15;11(1):20553.
- T Liechti, F Mair, et al. An updated guide for the perplexed: cytometry in the high-dimensional era. Nat Immunol. 2021 Oct;22(10):1190-1197.
- F Mair, AJ Tyznik. High-Dimensional Immunophenotyping with Fluorescence-Based Cytometry: A Practical Guidebook. Methods Mol Biol. 2019:2032:1-29.
- L Ferrer-Font, KM Price, et al. Panel Design and Optimization for High-Dimensional Immunophenotyping Assays Using Spectral Flow Cytometry. Curr Protoc Cytom. 2020 Mar;92(1):e70.
- TM Ashhurst, et al. High-Dimensional Fluorescence Cytometry. Curr Protoc Immunol. 2017 Nov 1:119:5.8.1-5.8.38.
ブログ著者について
 |
|
|
Frédéric Duval博士は、Maisonneuve-Rosemont病院研究センターのフローサイトメトリーコアマネージャーです。 |
Sari Gezzar-Dandashi氏はMontreal大学で分子生物学を専攻する博士課程5年生で、Maisonneuve-Rosemont病院研究センターのHugo Wurtele博士とElliot Drobetsky博士の研究室で研究を行っています。卵巣がん細胞株のサブ集団のDNA損傷誘発剤に対する様々な応答を、細胞が発現する細胞表面マーカーに基づき検出・定量するために、フローサイトメトリーを使用しています。 |
