
定量的miRNAプロファイリング:最適なRNAシーケンシング法を選ぶには?
RNAシーケンシング(RNA-seq)は、生体サンプル中のRNA分子の探索や定量化をするために、次世代シーケンシング(NGS)を用いて幅広い分野で使用されている手法です。
なぜRNA-seqを行うのか?
RNA-seqは、トランスクリプトミクスのアプローチに革命をもたらし、マイクロアレイやEST(expressed sequence tag、発現配列タグ)シーケンシングに比べて、ハイスループットで正確かつ高いコスト効率のRNA解析を実現します[1](表1)。
| パラメーター | DNAマイクロアレイ | ESTシーケンシング | RNA-seq |
| 検出方法 | ハイブリダイゼーション | サンガーシーケンシング | NGS(next generation sequencing) |
| 感度 | 高感度であるが、制限あり—発現レベルの変化を最大10,000倍まで検出 | 制限あり—small RNA(<500bp)の解析はできない。RNA特異的アイソフォームの読み取り・マッピングは不可能な場合がある。 | 高感度—シーケンス深度を高くとることで、1塩基レベルの分解能で低発現RNAを検出可能 |
| 必要なRNA量 | 中程度~多い—1~10ng | 多い—cDNAライブラリーを必要とする | 少ない—シングルセル解析も可能 |
| コスト | 中程度 | 中程度~高い—大半がcDNAライブラリーの構築とサンガーシーケンシングコストに関連 | 低い |
RNA-seqの技術は、基礎研究や臨床研究のアプリケーションに広く利用されています(図1)。
主な用途:
- すべてのコーディングRNA、ノンコーディングRNA、スプライス部位、アイソフォーム、オープンリーディングフレームの同定による、ゲノムの機能アノテーション
- RNA発現量の定量解析・発現プロファイリング
- 一塩基多型の検出
- RNA編集の研究—RNA転写後修飾(PTM:posttranscriptional modification)の研究
特殊なRNA-seqセットアップをすることで、例えばコーディングRNA(mRNAシーケンシング・エクソームキャプチャーシーケンシング)やリボソームRNA(リボソームプロファイリング)等のRNAのサブセットに焦点を当てることができます。RNAを検出する感度が向上したことで、単一細胞のシーケンシング法(シングルセルRNA-seq)が開発され、組織の複雑性や不均質な細胞集団を単一細胞レベルで研究するための非常に重要なツールとなっています。
RNA-seqは、RNA構造の決定(SHAPE、SHAPE:selective 2′-hydroxyl acylation analyzed by primer extension)や、RNA-RNAとRNA-タンパク質の相互作用部位のマッピング(CLIP、CLIP:Cross-linking immunoprecipitation)を目的に開発されました。
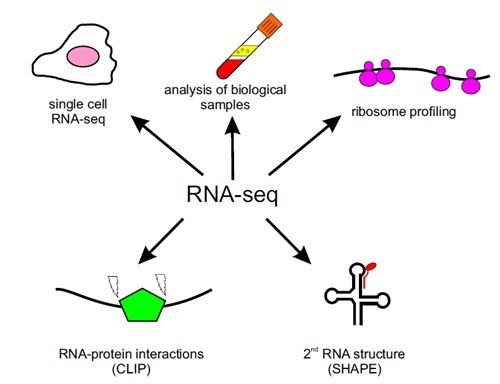
図1. 基礎研究および臨床研究におけるRNA-seq技術のアプリケーション例
small RNA-seqの課題
2008年頃に開発されたRNA-seqは、当初、ポリA配列を有するmRNA(メッセンジャーRNA)に対して適用されました[2-4]。標準的なRNA-seq(「long RNA-seq」)は、mRNAや長鎖ノンコーディングRNAのシーケンシングに使用されます。RNA-seqは、RNAの単離とそれに続く適切な鎖長30~400bpとなるようなRNAの断片化とcDNAへの逆転写に基づいた技術です。一方で、成熟したmicroRNA(miRNA)のような~約22塩基のsmall RNAは、標準的なRNA-seqプロトコールにはあまりにも鎖長が短すぎます。miRNAは鎖長が短いことから、バイアスがなく効率の高い逆転写やPCR(ポリメラーゼ連鎖反応)増幅を実現するには、ライブラリー調製時に伸長(RNAライゲーションやポリAテーリング等)という追加的なステップが必要となります(「small RNA-seq」、図2)。特に、血中の循環セルフリーmiRNAの検査等のRNA量が少ない生体由来サンプルでは、適切な正規化と再現性の評価が求められます。Giraldezら[5]は、Nature Biotechnologyにおいて、9種類の異なるライブラリー調製法の評価試験を行い、それらの再現性、精度、miRNAの編集を実施した場合の検出能を評価しました。
評価試験は、次の3つのリファレンスサンプルを用いて行われました。
1) 等モル比の1,000を超える合成small RNAプール
2) 異なるモル比に調製された合成small RNAプール
3) ヒトの血液から単離されたsmall RNAプール
9組の独立した研究グループが、複製物を調製しライブラリーを解析した結果、364という膨大な数のRNA-seqデータセットが得られました。
small RNA-seqの様々な方法
small RNA-seq実験を行うために何通りかの市販キットが開発され、研究グループらはいくつかの実験手法を考案しています。現在利用されている手法の大半で、3'および5'末端のアダプターライゲーションを必要とします。アダプターライゲーションは、増幅RNAの品質と選択性に直接影響を与える重要なステップです。ライゲーションステップで使用されるRNAリガーゼ(Rnl1およびRnl2)には、特定のRNAサブセットの過剰発現および発現低下をもたらすことになる配列決定バイアスおよび構造的バイアスが存在することが示されています[6,7]。small RNA-seqに関する現在のプロトコールと解析方法は、2種類のカテゴリに分けることができます。すなわち、インバリアントな末端を持つアダプターを利用する方法(アダプター配列はあらかじめ厳密に決められています)、および、ライゲーション末端に4塩基の縮重ヌクレオチド(縮重塩基)を含むアダプター(4Nアダプター)を利用する方法です(図2)。
Giraldezらは、インバリアント末端を用いて、TruSeq(Illumina社)、NEBNext(New England BioLabs社)、CleanTag(Trilink Biotech社)の3つの市販キットを評価しました。また、4Nアダプターについては、市販のNEXTflex(Bioo Scientific社)、2015年にPingら[8]によって公開されたプロトコール、および4組の研究グループが考案したプロトコールの計6種類のプロトコールを評価しました。
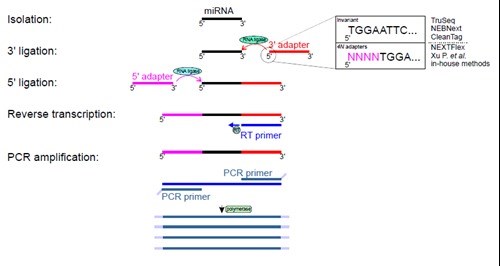
図2. small RNA-seqの実験手順。単離されたsmall RNAは、3'および5'末端アダプターで2回のライゲーション反応を行う。このライゲーション反応は2種類のRNAリガーゼによって触媒される。得られた産物を逆転写の鋳型とし、DNAフラグメントを数サイクルのPCRで増幅させる。増幅されたDNA産物についてシーケンシングを行う。
Giraldezら[5]は、3’末端アダプターおよび5’末端アダプターのライゲーション末端にインバリアント末端アダプターを使用する方法、または4塩基の縮重ヌクレオチドを挿入したアダプターを使用する方法のいずれかを採用した9種類のプロトコールを比較しました。図では単純化して3'末端アダプターのバリアントのみを示しています。
1. シーケンスバイアスは非常に重要な問題です
Giraldezら[5]は、現在使用されているsmall RNA-seqのプロトコールにはシーケンスバイアスの問題があると報告しています。異なるプロトコールで得られた結果間に認められるシーケンスバイアスは、同一のプロトコールを使用して異なる研究グループが作成したデータセット間に認められるシーケンスバイアスよりも有意に大きく、RNA-seqのプロトコールの選択が、得られるデータの品質に大きな影響を与えることを示唆しています。異なるプロトコールで調製したライブラリーを使用して解析された類似する生物学的現象のsmall RNA-seqの結果を比較する際、このシーケンスバイアスに留意する必要があります。4Nアダプターを使用する方法は、インバリアントアダプターを使用する方法に比べてシーケンスバイアスが小さいことから(プロトコールによって2~20倍程度異なる)、推奨される手法です。4Nアダプターを使用するプロトコールではシーケンスバイアスが小さくなることから、シーケンシングの深度が低い場合でも、多くの種類のsmall-RNA鎖を検出する(サンプルのカバー率を高くする)場合により良好な結果が得られることを示唆しています。
2. RNA-seqプロトコールにおける精度および再現性
あらかじめ定められた合成small-RNAのプールを使用することにより、異なる研究グループ間で得られた結果について、すべてのプロトコールの精度および再現性の比較が実施されました。観察されたmiRNA存在量の比は、予想された比と非常に近似していました。この近似は、すべてのプロトコールが正確な相対的miRNA存在量を測定するための精度を有していたことを意味しています。再現性は、合成small RNAおよび生体由来small RNAサンプルの統計的パラメーター(変動係数、四分位分散係数)により評価されました。詳細な結果は論文中の補足資料に要約されています(評価に用いられたRNA配列の変動係数の大半は20%未満の値を示しています)[5]。
3. 研究グループが考案した4Nアダプターを用いるプロトコールでmiRNAを検出するケースが最も信頼性が高い
アデノシン残基からイノシン残基への脱アミノ化(A-to-I編集)は、miRNA前駆体のサブセットで発生することがあります。これは、その後のプロセシングや生物学的活性に影響を与えます[9]。したがって、miRNAがA-to-I編集されていることを正確に検出できる適切なRNA-seq法を研究者が採用することが重要となります。Giraldezら[5]は、異なるモル比に調製した編集済みの合成small RNAおよび未編集の合成small RNAをリファレンスサンプルに使用しましたが、9種類のプロトコールのうち3種類がA-to-I miRNA編集を検出できると評価しました。評価に用いたプロトコールはすべて良好な特異性を示し(>99%)、サンプル中の編集済みのRNAおよび未編集のRNAを確実に検出し識別することができました。しかし、実際のRNA編集の程度を正確に検出するという点では、研究グループが考案した4Nアダプターを使用するプロトコールの方が市販のインバリアントアダプターを使用するプロトコールよりも優れたパフォーマンスを示しました。このことは、4Nアダプターを使用するプロトコールの方がよりシーケンスバイアスが小さく、シーケンスのカバレッジを最大化するため、インバリアントアダプターを使用するプロトコールよりもパフォーマンスが優れている可能性が高いことが考えられます。
small RNA-seqの解析にはさらなる改善が求められます
疑いの余地もなく、この包括的な研究は、研究者がsmall RNA-seqのプロトコールや実験デザインの詳細について、より踏み込んでフォローアップをする一助となります。small RNA-seqは、long RNA-seqと比較すると、依然として大きなシーケンスバイアスを示します。重要なことに、4Nアダプターを使用したsmall RNA-seq解析の最近の研究では、シーケンスバイアスを小さく抑えられているようでした。検証した4Nアダプターを使用したプロトコールで、異なる品質のデータセットが生成されたということは、small RNAに関する実験を行う際に、ライゲーションや温度等の追加パラメーターを考慮する必要があることを意味し、この問題について言及する価値があったといえます。
参考:
投稿者:Karolina Szczesna博士、プロテインテック シニアプロダクトマネージャー兼テクニカルサポート
参考文献
- Wang, Z., M. Gerstein, and M. Snyder, RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 2009. 10(1): p. 57-63.
- Mortazavi, A., et al., Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 2008. 5(7): p. 621-8.
- Nagalakshmi, U., et al., The transcriptional landscape of the yeast genome defined by RNA sequencing. Science, 2008. 320(5881): p. 1344-9.
- Wilhelm, B.T., et al., Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature, 2008. 453(7199): p. 1239-43.
- Giraldez, M.D., et al., Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat Biotechnol, 2018. 36(8): p. 746-757.
- Jayaprakash, A.D., et al., Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res, 2011. 39(21): p. e141.
- Zhuang, F., et al., Structural bias in T4 RNA ligase-mediated 3'-adapter ligation. Nucleic Acids Res, 2012. 40(7): p. e54.
- Ping, X., et al., An improved protocol for small library construction using High Definition adapters. Methods Next-Generation Seq., 2015(2): p. 1-10.
- Yang, W., et al., Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol, 2006. 13(1): p. 13-21.