
フローサイトメトリーの蛍光補正
Shweta Puntambekar博士のウェビナー「Mastering Flow Cytometry: Best Practices and Tips」より
フローサイトメトリーの蛍光補正(Compensation)とは?
プロテインテックは2023年4月11日に、Shweta Puntambekar博士によるワークショップウェビナー「Mastering Flow Cytometry: Best Practices and Tips」を開催しました。本稿では、このウェビナーのフローサイトメトリーの蛍光補正に関する解説を抜粋してご紹介します。
※本記事は以下のウェビナー「Mastering Flow Cytometry: Best Practices and Tips」の内容をもとに内容を構成したものです。判読性向上のため一部の内容を編集しています。
https://www.ptglab.com/workshops/mastering-flow-cytometry-best-practices-and-tips/
(視聴者向け割引特典は現在はご利用いただけません)
スペクトルのオーバーラップ(重複)とは?
レーザーを照射して蛍光を解析する典型的な例として、サンプルを染色してFITCによる蛍光とPEによる蛍光を観察する実験を行ったと仮定します。装置には、FITCやPEを励起できるレーザーが搭載されており、励起された色素はそれぞれ固有の蛍光スペクトルを発し、そのシグナルのデータを解析するとします。
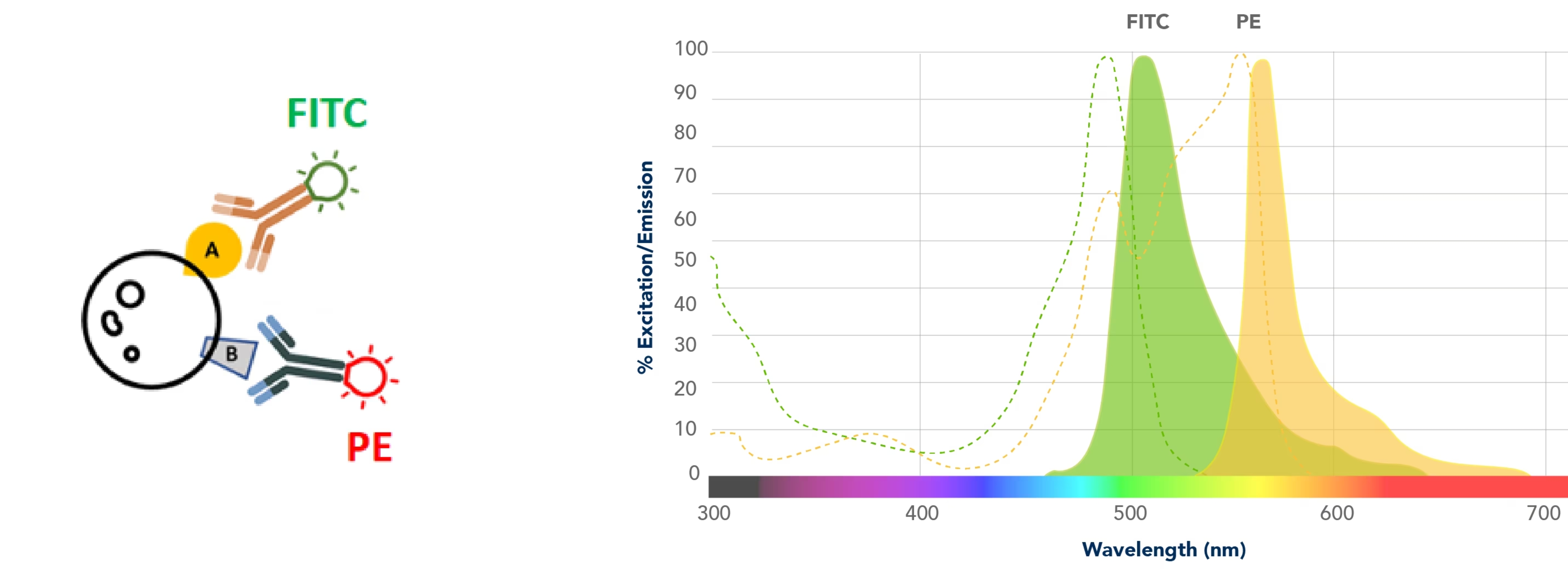 |
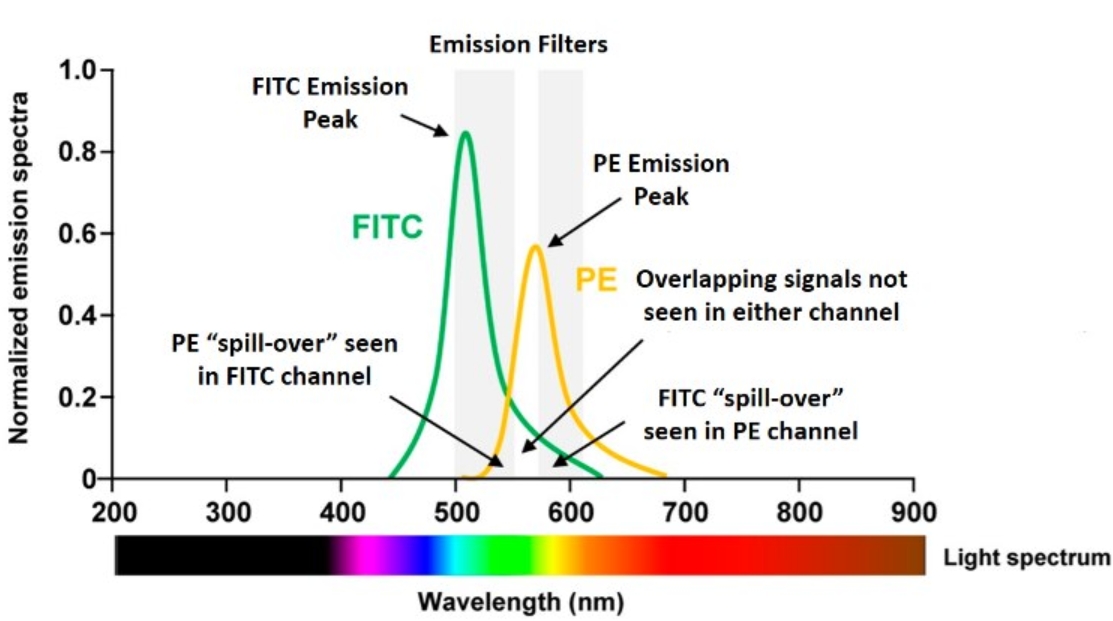
フローサイトメーターには非常に優れたフィルターが搭載されています。蛍光検出では常に何らかのスペクトルのオーバーラップ(重複)が存在しますが、こうした専用のフィルター類が非特異的なオーバーラップシグナルの多くを除去します。しかし、フィルターを使用したとしても蛍光色素の発光波長域には重なりが多く、オーバーラップの問題を完全に解決できない点はご理解いただけるかと思います。下図の左側の蛍光スペクトルをご覧ください。FITCスペクトルにFITCフィルターを通過するPE由来のスペクトルがオーバーラップしており、FITC検出器にPE由来の蛍光が漏れ込んでいる(spill-over)状態になっています。また、右側のPEスペクトルにPEフィルターを通過するFITC由来のスペクトルがオーバーラップしています。
 |
得られたデータのFITC+/PE+細胞集団が実際に陽性細胞であり、スペクトルのオーバーラップに起因する蛍光の漏れ込みではないことを確認するにはどうすれば良いでしょうか?フローサイトメトリーでは、1種類の蛍光色素で染色したサンプル(単染色コントロール)を使用してスペクトルのオーバーラップによる漏れ込みを補正することができます。まさに、この点はフローサイトメトリーの大きな長所といえます。比較的単純な実験系を例に挙げると、未染色の細胞やビーズ等を用いて自家蛍光等のバックグラウンドを計測して、これをネガティブコントロールとします。そして、1種類の蛍光色素標識抗体で染色したコントロールサンプルを、使用する蛍光色素の数だけ用意して計測します。例えば、FITC標識抗体1種類だけで染色した細胞と、PE標識抗体1種類だけで染色した細胞を用意します。
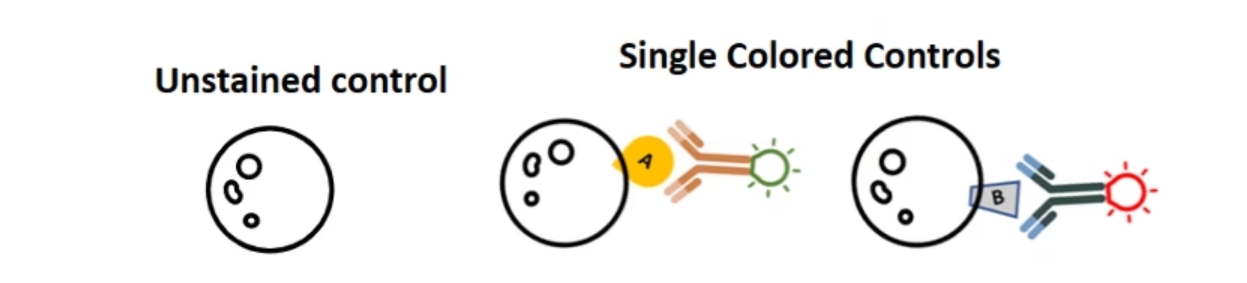 |
ユーザーがよく犯す間違いの1つに、単染色コントロールサンプルは作製するものの、未染色のコントロールサンプルを実験系に含めないことが挙げられます。ネガティブコントロールの基準を定義するには、必ず未染色の細胞やビーズが必要になります。単染色コントロールサンプルを測定して得られるデータをネガティブコントロールとして使用することはできません。
蛍光補正(Compensation)
ここでは例として、FITC標識単染色コントロールサンプルを測定したとします。このサンプルはFITCしか標識されておらず、PEの蛍光は認められないはずです。しかし、サンプルを測定すると、PE+集団が存在するかのようなデータが得られます。このFITC+/PE+集団は実在しない集団です。このアーチファクトは、FITC由来の蛍光がPEチャンネルに漏れ込んで生じたデータです。そのため、PEチャンネルに漏れ込んだFITCの蛍光を差し引く必要があります。具体的にはこのPEチャンネルに漏れ込んだFITC由来のシグナルを蛍光補正によりゼロレベルに修正します。補正を行った結果、このサンプルでは偽陽性の問題が解消され、FITC由来の蛍光だけが正しく検出されるようになります。
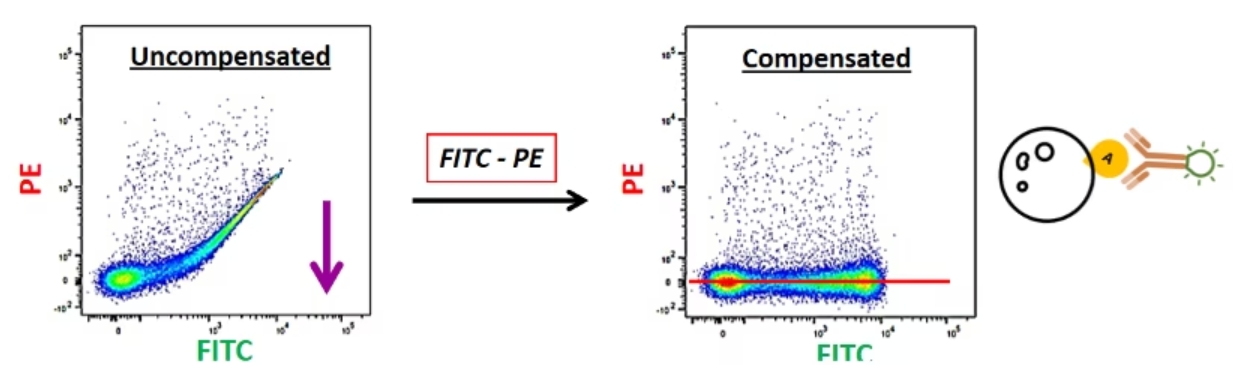 |
同様に、PE標識単染色コントロールサンプルを測定してFITC+集団が認められた場合、このFITC+集団は実在しないアーチファクトです。FITCチャンネルで検出されるシグナルは、PE由来のシグナルです。そのため、FITCチャンネルからPE由来の蛍光を減じる蛍光補正を適用すると、すべての蛍光シグナルはFITC-/PE+になり、FITCチャンネルのPE由来の漏れ込みシグナルはなくなります。
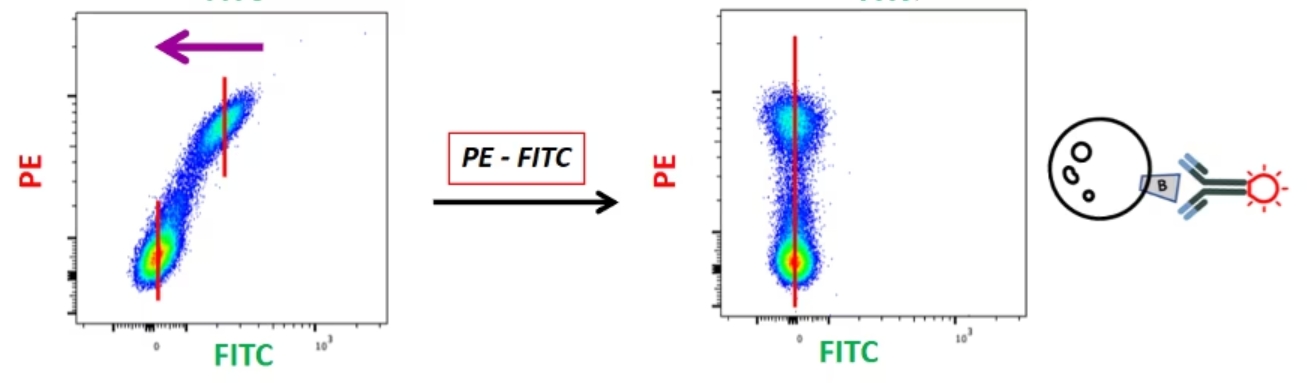 |
この漏れ込みシグナルの減算に利用するのが、いわゆる補正コントロールです。蛍光補正で理解しておく必要があるのは、「『FITC-PE(FITC検出器に漏れ込んだPE由来の蛍光を減じる補正)』と『PE-FITC(PE検出器に漏れ込んだFITC由来の蛍光を減じる補正)』は2つの別々の補正である」ということです。この例では、2種類の異なるレーザー光と、2種類の異なる漏れ込みを対象としていることになります。
蛍光補正のポイント
単染色コントロールには何を使用するべきでしょうか?私の経験則では、ポジティブ集団とネガティブ集団のピークが明瞭に分離しているコントロールサンプルは、優れた単染色コントロールであるといえます。そのため、常にこの判断基準に基づいて検討を行っています。
多くの研究者は細胞を使用するのを好みます。コントロールに細胞を使用する利点は当然のことながら、本試験と同じ細胞を使用すれば前方散乱光と自家蛍光の特性が一致するという点です。そのため、実際のサンプルを使用してどのようなデータが得られるか、あるいは本試験でオーバーラップに起因する蛍光の漏れ込みや蛍光補正等の設定にどの程度影響を及ぼすのかを確認することができます。細胞を使用するデメリットは、用意できるサンプル量が少なく、貴重なサンプルが余計に消費されてしまう点です。実際の測定に使用する細胞と単染色コントロールに使用する細胞を常に確保できるとは限りません。また、大きなデメリットの1つに生体由来のサンプルは明瞭なピークを得られないという点が挙げられます。生体由来サンプルを用いた場合、大抵はスメアな(分布がブロードな)データが得られます。細胞集団の明確なポジティブピークやネガティブピークを得るのは困難です。そのため、ピークが明瞭に分離する蛍光補正コントロールが極めて重要となります。
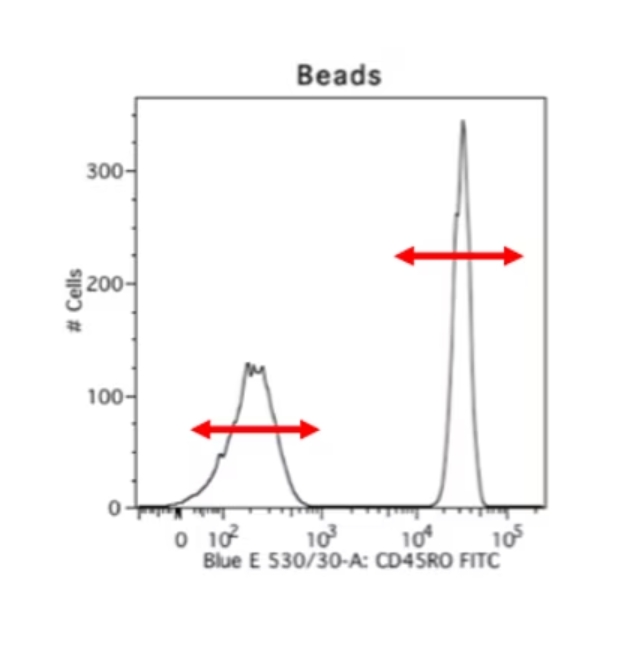
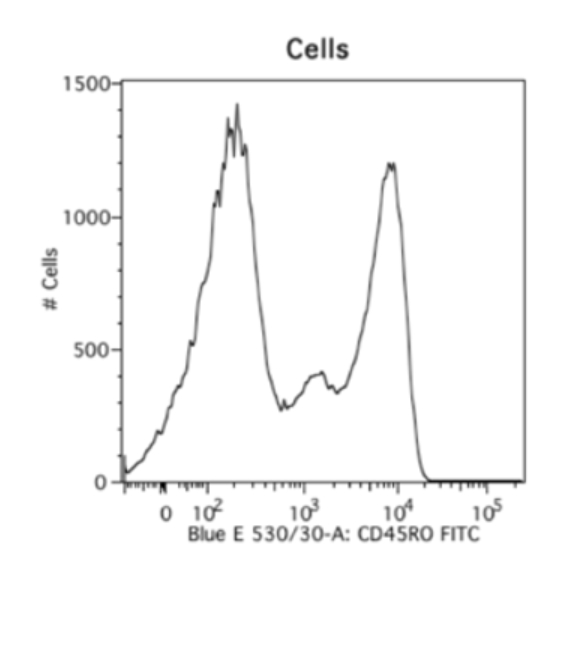
細胞等の生体試料の課題は、ビーズを用いる手法によってある程度解決できます。ビーズを用いる手法は大変便利で好ましい手法です。蛍光補正コントロールの染色再現性が高く、サンプル細胞を実際に観察したいマーカー染色だけに使用できるのに加え、ポジティブピークとネガティブピークが極めて良好に分離します。以下には、同一の抗体を用いて染色した生体由来の細胞サンプルと、ビーズをサンプルとして測定した比較データの例を示しました。生体試料を測定した右のデータは不明瞭なスメアが認められ、ネガティブシグナルの終了ポイントとポジティブシグナルの開始ポイントが不明確になっています。しかし、ビーズを使用して測定した左のデータは、明確なポジティブピークとネガティブピークが認められます。そのため、私は常に蛍光補正を実施する際にビーズを使用します。ビーズを使用するデメリットは、自家蛍光のレベルや、前方散乱光、側方散乱光のパラメータが細胞サンプルと異なるという点です。
  |
蛍光補正の設定をする際は、ポジティブピークの出現領域等を考慮して使用する検出器の設定も行います。電圧を高くすればシグナル強度も強くなり、オーバーラップに起因する蛍光の漏れ込みの影響が大きくなります。蛍光補正用のパラメータは、PEやFITC等で実際に染色した細胞を測定する際の電圧をもとに設定します。前方散乱光や側方散乱光は蛍光補正の設定に含まれないため、ビーズを使用して蛍光補正の設定を適用した後でも、蛍光補正とは別に変更することが可能です。散乱光は細胞のサイズ・粒度等を測定するためのものです。散乱光の設定は変更可能ですが、その他のパラメータや、目的のターゲットを測定する蛍光検出器の設定は変更しないでください。
Puntambekar博士はNeurocrine BiosciencesのSenior Scientistとして現職に就く前は、インディアナ大学医学部スターク神経科学研究所のAssistant Research Professor兼フローサイトメトリーコア施設のScientific Managerを務めていました。
