
レット症候群—女性における精神遅滞の原因第二位
1966年、レット症候群が初めて臨床上の問題として報告されました。
レット症候群について
レット症候群(RTT:Rett syndrome)は、出生女児の1万人に1人が発症する精神遅滞であり、女児における精神遅滞の原因の第二位を占めています。1999年、Zoghbi博士の研究室で、レット症候群の変異遺伝子が同定されました。典型的なRTTの95%は、MeCP2変異を原因としています(1)。
MeCP2は、様々な組織で広く発現する核タンパク質であり、成熟した神経系のニューロンに最も多く存在します(1、図1)。
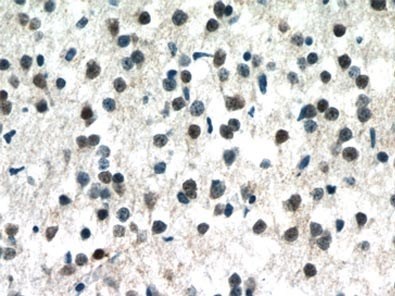
図1. MECP2抗体(カタログ番号:10861-1-AP、希釈倍率1:50)を使用したパラフィン包埋ヒト脳組織スライドの免疫組織化学染色(40倍レンズを使用)。
MeCP2の機能は完全には解明されていませんが、特定の遺伝子のサイレンシングや活性化を介して遺伝子発現を制御している、あるいは、より広範な転写プロセスの制御によって遺伝子発現を制御しているのではないかと考えられています(2)。RTTは、エピジェネティクスと関連が認められた最初の神経発達障害です。それ以降、CDKL5やFOXG1遺伝子もまた、レット症候群様の精神遅延との関連が認められています(3,13)。
レット症候群の臨床的特徴
RTTは、出生前および周産期に異常の認められない、産後に起こる進行性疾患です。レット症候群の患者であっても、生後6か月から18か月の間は然るべき脳の発達が認められ、この期間は発達停滞期と呼ばれています。この期間、患者女児は、正常な神経発達、運動機能、コミュニケーション能力を有します。しかし、その後、発達の退行が生じます(4,5)。
この疾病の退行期の進行期間は、1歳から4歳の間に生じ、患者女児は会話ができなくなり、手の合目的運動が消失します。患者は高い頻度で、主に対人接触の減衰に関連した自閉症様の特徴を示します。また、小頭症を原因とする頭部の成長速度の鈍化が認められます。神経学的評価では、脳波検査で大脳皮質の過剰興奮が認められ、発達の喪失を示します。
4歳から7歳の間に起こる第3期(仮性安定期)では、患者女児は人に関心を示すようになりますが、歯ぎしり、発作的な叫び声、重度の脊椎側彎症、身体の成長遅延、抑うつ気分、夜泣き、夜間に笑い出すといった異常を示すようになります。てんかんが認められるようになるのもこの時期です。一般的に手絞り、手洗いの動作の他に、手を叩く・拍手をする、あるいは握りしめる動作といった、手の常同運動が認められます。アイコンタクトは、当初低下していますが何らかの形で回復し、小児は要求を表現するため、一般的にアイ・ポインティング(eye-pointing)で人に関心を示したり喜んだりするようになる場合があります。
5歳から15歳以上で起こる晩期運動機能低下期には、患者女児は、呼吸機能障害による突然死のリスクが高くなる心臓異常(徐脈や頻脈)を発症するようになります。多くの場合、運動機能がなくなることで、硬化硬直(frozen rigidity)と呼ばれる状態に陥ります。しかし、中には歩行能力を失うことなく、生涯ステージ3を維持する患者女児もいます(図2に概要を記載)。
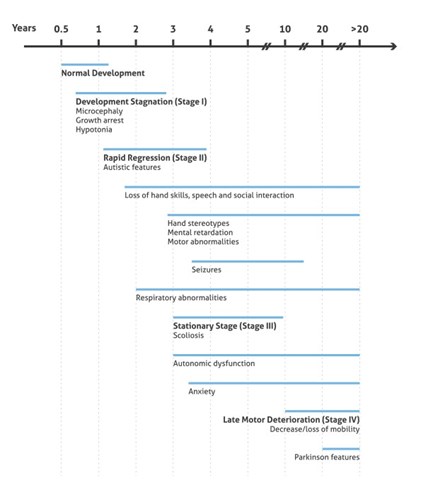
図2. RTTの臨床表現型の発症と進行の図表((3)を基に作成)。
レット症候群患者に認められる異常と神経病理
RTTに罹患した患者の脳重量は、同年齢の対照健常者の脳に比べて有意に小さい特徴を示しますが、年齢に伴う脳重量の有意な減少は認められませんでした(6)。ニューロンがより小さく密集するため、脳の容積が減少します。
主要な変化は、前頭領域、後側頭部領域、後頭後部領域と関連しています。神経病理学的には、アセチルコリン(7,8)、ドーパミン(9)(図3)、セロトニン(10)、グルタミン酸(11)、神経成長因子(12)等、脳脊髄液にも病態が認められます。
また、MAP2(microtubule-associated protein 2、微小管結合タンパク質2)(図4)の発現は、樹状突起形成時におけるMeCP2の関与にあわせて、ニューロンの可視化に寄与します。
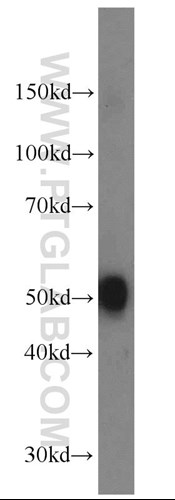
図3. SDS-PAGE後、DOPAデカルボキシラーゼ抗体(カタログ番号:10166-1-AP、希釈倍率1:1000、室温で1.5時間インキュベート)を使用したマウス脳組織のウェスタンブロット。
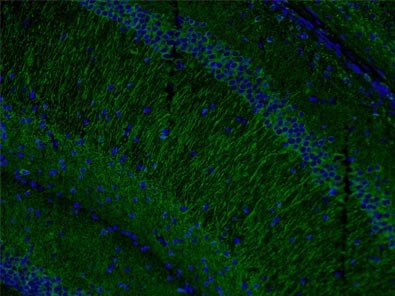
図4. MAP2抗体(カタログ番号:17490-1-AP、希釈倍率1:50)と、Alexa Fluor 488標識AffiniPureヤギ抗ウサギIgG(H+L)抗体を使用した、マウス脳組織の免疫蛍光染色解析像(4% PFAで固定)。
レット症候群の動物モデル
マウスの脳はヒトの脳と類似した構造をしているため、マウスモデルは多くの疾病のメカニズムや病態の解明に役立っています。
未だにレット症候群に有効な治療法は存在しません。MeCP2ノックアウトマウスは、ヒトのレット症候群に類似した様々な生理学的および神経学的異常を有しており、レット症候群の異常を標的とした新しい臨床治療法を検討するためのモデルとして使用できます。
著者:Karolina Szczesna(生物医学博士)
プロテインテック シニアプロダクトマネージャー
参考文献:
- R.E. Amir, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999 Oct;23(2):185-8.
- M. Chahrour, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008 May 30;320(5880):1224-9.
- M. Chahrour, et al. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007 Nov 8;56(3):422-37.
- B. Hagberg. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8(2):61-5.
- E. Trevathan, et al. The clinical recognition and differential diagnosis of Rett syndrome. J Child Neurol. 1988;3 Suppl:S6-16.
- D.D. Armstrong. Neuropathology of Rett syndrome. J Child Neurol. 2005 Sep;20(9):747-53.
- G.L. Wenk. Rett syndrome: neurobiological changes underlying specific symptoms. Prog Neurobiol. 1997 Mar;51(4):383-91.
- G.L. Wenk, et al. Choline acetyltransferase activity and vesamicol binding in Rett syndrome and in rats with nucleus basalis lesions. Neuroscience. 1996 Jul;73(1):79-84.
- H.Y. Zoghbi, et al. Reduction of biogenic amine levels in the Rett syndrome. N Engl J Med. 1985 Oct 10;313(15):921-4.
- M. Segawa, et al. Polysomnography in the Rett syndrome. Brain Dev. 1992 May;14 Suppl:S46-54.
- R. Lappalainen, et al. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol. 1996 Oct;15(3):213-6.
- R. Lappalainen, et al. Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J Child Neurol. 1996 Jul;11(4):296-300.
- F. Ariani, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008 Jul;83(1):89-93.