
ゲノムDNA損傷の修復法
DNA損傷の修復機構:加齢に伴う疾患・腫瘍形成の防止機構
DNA損傷の修復機構
リンクをクリックすると移動します
| I. 直接修復(Direct reversal) | b)ヌクレオチド除去修復(NER) |
| II. ミスマッチ修復(MMR) | a)非相同末端結合(NHEJ) |
| III. 一本鎖切断(SSB)の修復 | b)マイクロホモロジー媒介末端結合(MMEJ) |
| IV. 二本鎖切断(DSB)の修復 | c)相同組換え(HR) |
| a)塩基除去修復(BER) | DNA損傷修復機構が機能しなくなったら? |
プロテインテックの特集:「DNA損傷修復マーカー」はこちら をご覧ください。 |
未修復のDNA損傷は、突然変異誘発や細胞死につながります。筋細胞や神経細胞にDNA損傷が蓄積すると、変性疾患を引き起こす可能性があります(1)。日々、細胞は様々なDNA損傷因子に曝されています(2)。DNA修復の主な機構は、一体どのようなものでしょうか?(図1)
プロテインテックの関連ブログ:失われた塩基対:DNA損傷
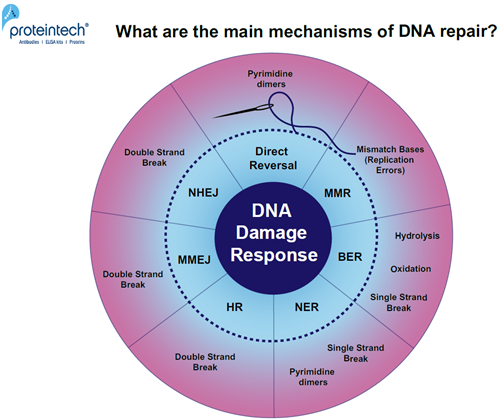
図1. DNA損傷特異的修復経路の概要。応答メカニズム:直接修復(Direct reversal)、MMR(MisMatch Repair:ミスマッチ修復)、BER(Base Excision Repair:塩基除去修復)、NER(Nucleotide Excision Repair:ヌクレオチド除去修復)、HR(Homologous Recombination:相同組換え)、MMEJ(Microhomology-Mediated End Joining:マイクロホモロジー媒介末端結合)、NHEJ(Non-Homologous End Joining:非相同末端結合)。
DNA損傷の修復機構
細胞は、DNA損傷部位を認識して目印を付け、DNA損傷応答で働く因子を誘引します。細胞周期チェックポイントの活性化、つまり細胞周期停止期間は、修復機構が働く時間です(図2)。DNA修復は、細胞周期のいくつかの段階で起こりますが、細胞はその大半がG1/S期とG2/M期に停止します。複製エラーにより損傷が生じた場合は、S期や有糸分裂後期であっても細胞周期は停止します。
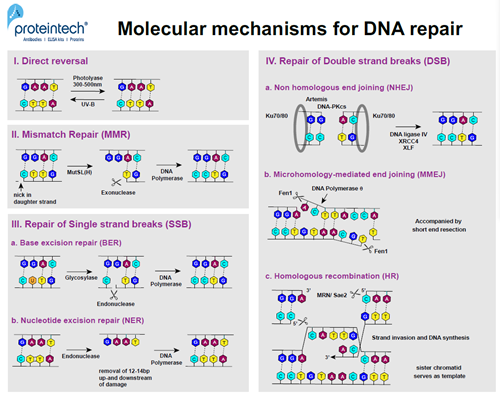
図2. 分子レベルでのDNA修復機構
I. 直接修復(Direct reversal)
ノーベル賞を受賞したAziz Sancar氏は、大腸菌の酵素フォトリアーゼを最初に発見した人物です(3)。紫外線曝露に反応し、2つの隣接するピリミジン残基が共有結合を形成し、結果としてピリミジン二量体(Py-Py)が生じます。フォトリアーゼは、300~500nmの波長で活性化されます。この酵素は、ホスホジエステル結合を壊すことなく、サブナノ秒以内にPy-Py二量体の共有結合を開裂する働きがあります(4)。現在では、直接修復は原核生物と真核生物の両方における修復機構であることが判明していますが、ヒトはこれにあてはまらず、代わりにNERが用いられます(図2;I)。
II. ミスマッチ修復(MMR)
ミスマッチ修復(MMR)は、Paul Modrich氏(2015年ノーベル賞受賞)による大きな発見の1つです。MMRは、複製の過程でDNAの親鎖と新しく合成された「娘」鎖を識別する細胞の能力に基づいています。バクテリアでは、DNAメチル化は親鎖のみに認められ、鋳型DNAを特定する校正機構のマーカーとして働き、「娘」鎖のミスマッチ塩基の探索に用いられます。真核生物では、DNAメチル化がマーカーとして働くエビデンスは存在せず、リーディング鎖とラギング鎖の複製中に生じる損傷が、校正機構のリクルートメントとして働きます。MutSとMutL(大腸菌ではMutHも関与します)、2種類の二量体の協調する働きにより、「娘」鎖のミスマッチ塩基が認識され、ミスマッチ塩基から最大1kbまでの範囲の特異的部位に切れ目が形成されます。エキソヌクレアーゼがその切断部位からミスマッチ部位までのDNAを除去し、DNAポリメラーゼδ(原核生物の場合はポリメラーゼⅢ)が除去された領域を最終的に再合成します(図2;Ⅱ)。
III. 一本鎖切断(SSB)の修復
一本鎖切断が起こると、損傷鎖は最初にATR(Ataxia Telangiectasia and Rad3-related:毛細血管拡張性運動失調症およびRad3関連キナーゼ)とRPA(Replication Protein A:複製タンパク質A)と結合し、マークされます。ATRは、自己リン酸化により活性化し、ATRIP(ATR Interacting Protein:ATR結合タンパク質)と二量体を形成します。それに続いて、9-1-1複合体(Rad9、Hus1、Rad1サブユニットで構成される)を一本鎖DNA切断(SSB)の端にリクルートします(5)。ATRは、Chk1(Checkpoint Kinase 1:チェックポイントキナーゼ1)をリン酸化し、Chk1によるCdc25の阻害を介して最終的に細胞周期の停止に至ります(6)。その後、SSBは、以下に示す2つの代替機構で修復されます。
a)塩基除去修復(BER)
塩基除去修復(BER)は、Tomas Lindahl氏によって初めて発表された機構で、主に単一ヌクレオチドの損傷に応答して生じます(7)。グリコシラーゼは、各塩基対の状態を評価するために各DNA鎖の塩基に沿って移動し、塩基をフリップアウトします。間違った塩基が発見されると、その塩基は除去され、APエンドヌクレアーゼがDNA骨格のホスホジエステル結合を切断します。続いて、生じたギャップをDNAポリメラーゼが正しいヌクレオチドで埋めます。この修復機構は、脱プリン塩基(プリン塩基の消失)の修復にも用いられますが、その場合、グリコシラーゼを必要としません(図2;Ⅲa)。
b)ヌクレオチド除去修復(NER)
ヘテロ二量体DDB(DNA Damage-Binding:DNA損傷結合)は、より規模の大きなDNA損傷(例:ピリミジン二量体化)の認識に関係しています。それには、コアヒストンであるH2A、H3、H4のユビキチン化とXPCが関与しています。こうした因子の働きによってヌクレオチド除去修復(NER)機構が活性化し、損傷したDNA領域周辺の12~24ヌクレオチドの除去が開始されます。続いて、RFC(Replication Factor C:複製因子C)とPCNA(Proliferating Cell Nuclear Antigen:増殖細胞核抗原)がDNAに結合することで、DNAポリメラーゼ(δ、ε、κ)によってギャップを埋めることができる状態になります(図2;Ⅲb)。
IV. 二本鎖切断(DSB)の修復
ATM(Ataxia-Telangiectasia-Mutated:毛細血管拡張性運動失調症変異タンパク質)は、二本鎖切断(DSB)によるDNA損傷応答における重要な役割を担っています。不活性型ATMは通常、多量体として存在し、DNA損傷時の自己リン酸化と活性化により解離します(5)。ATMは、続いてDSB部位にリクルートされ、そこでH2AXヒストンバリアントをリン酸化します。H2AXからγH2AX(リン酸化H2AX)へのリン酸化は迅速な反応であり、そのため優れたDNA損傷マーカーとして機能します。また、ATMはリン酸化によってChk2(Checkpoint Kinase 2:チェックポイントキナーゼ2)および/またはp53を活性化します。その結果、Cdc25が阻害されることにより細胞周期が停止し、続いてDNA損傷の修復あるいはアポトーシスに至ります。細胞周期の中でいつDSBが発生するかに応じて、以下の経路のいずれかがその修復に使用されます。
a)非相同末端結合(NHEJ)
Kuヘテロ二量体(Ku70-Ku80)は、DSB部の端に結合し、非相同末端結合(NHEJ)因子が接合するための環を形成します(8)。Kuは、配列に関係なくDNAに結合し、それは平滑末端や、5’突出末端、3’突出末端でも生じます。KuがDNAに結合すると、別のPI3K関連キナーゼであるDNA-PKcsをリクルートします。Artemisヌクレアーゼがリクルートされ、リン酸化されると、一本鎖DNAの突出末端が除去され、リガーゼⅣ、XRCC4、XLFによって2つのエンド末端が結合します。NHEJは非保存的なDNA修復経路であるため、通常は損傷領域の欠損が生じます(図2;Ⅳa)。
b)マイクロホモロジー媒介末端結合(MMEJ)
マイクロホモロジー媒介末端結合(MMEJ)の際、DSBの片側鎖上のマイクロホモロジー領域が識別され、突出一本鎖DNAはその配列に従って整列します。DNAポリメラーゼθは、マイクロホモロジー整列領域のアニーリングとライゲーションを促進します。Fen1ヌクレアーゼ(Flap Endonuclease 1:フラップ・エンドヌクレアーゼ1)が一本鎖DNAの突出部を除去し、DNAポリメラーゼθがその隙間を埋めます。この修復経路は常に切断部のDNA末端の短い削り込みを伴い、これによりDNA鎖中の塩基が欠失し、突然変異を引き起こします(図2;Ⅳb)。
c)相同組換え(HR)
相同組換え(HR)は、通常G2期に生じますが、他の2種類のDSB修復経路と比較してエラーが生じにくい傾向にあります。HRでは、姉妹染色分体の配列を鋳型として使用します。最初にMRN複合体(Mre11、Rad50、Nbs1)が切断部の端に結合します。次に、MRNとSae2は、DSB周辺の5’末端側をせん断し短い3’突出末端が生じます。RPAとRad51が一本鎖を覆い、核とタンパク質のフィラメント(ヌクレオプロテインフィラメント)を形成します。このフィラメントは相同領域を発見すると「姉妹」染色分体の無傷の二本鎖DNAに侵入します。続いて、RFC、PCNA、DNAポリメラーゼ(δ、ε、α)が侵入鎖の端に結合し、無傷の「姉妹」染色分体鎖を鋳型として使用し、消失した領域を合成します(図2;Ⅳc)。
DNA損傷修復機構が機能しなくなったら?
DNA損傷の蓄積は、一般的にがんや変性疾患との関連が認められ、通常は特定の細胞型や臓器に影響を及ぼします。これに対して、遺伝性疾患等の結果として、1種類以上のDNA修復タンパク質が機能を消失すると、すべての種類の細胞のDNA損傷修復の働きが同時に損なわれます。例えば、色素性乾皮症、コケイン症候群、硫黄欠乏性毛髪発育異常症等の遺伝性疾患は、ヌクレオチド除去経路の欠損を示します(9、10)。ヌクレオチド除去経路は、概して一本鎖切断、主にUV-Bによって生じるピリミジン二量体等の修復に関与しています。こうした疾患の患者は光感受性が高く、皮膚がんを発症しやすい傾向にあります。
その他の遺伝性疾患の例としてファンコニ貧血が挙げられますが、その疾患では相同組換え(HR)に関与する数種類のタンパク質が影響を受けている可能性があります。その症状には、発育異常や骨髄不全等があります。
さらに重い疾患となるのは、毛細血管拡張性運動失調症やSeckel症候群の原因となるATMやATRの突然変異です。こうした変異は、成長障害、神経変性、小頭症、高いがん発症リスクと関連しています。DNA損傷とその修復過程に関する多くの疑問は解決されておらず、将来的にDNA損傷の突然変異や機能異常の治療法を解明するうえで極めて重要となります。
プロテインテックの特集:「DNA損傷修復マーカー」はこちらをご覧ください
参考文献
1. Stein and Toiber (2017) “DNA damage and neurodegeneration: the unusual suspect”.
2. Lindahl and Barnes (2000) “Repair of endogenous DNA damage“.
4. Kao et al. (2005) “Direct observation of thymine dimer repair in DNA by photolyase”
5. Qiu et al. (2018) “ATR/CHK1 inhibitors and cancer therapy”.
6. Awasthi et al. (2015) “ATM and ATR signaling at a glance”.
7. Lindahl (2012) “My Journey to DNA Repair”
8. Frit et al (2019) “Plugged into the Ku-DNA hub: The NHEJ network”
9. Menck and Munford (2014) “DNA repair diseases: What do they tell us about cancer and aging?”
10. Torgovnick and Schumacher (2015) “DNA repair mechanisms in cancer development and therapy”